-
All topics
From our earlier studies, we know that liquids and gases are called fluids because of their ability to flow. The fluidity in both of these states is due to the fact that the molecules are free to move about. On the contrary, the constituent particles in solids have fixed positions and can only oscillate about their mean positions. This explains the rigidity in solids. These properties depend upon the nature of constituent particles and the binding forces operating between them. The correlation between structure and properties helps in the discovery of new solid materials with desired properties. For example, carbon nanotubes are new materials that have potential to provide material that are tougher than steel, lighter than aluminium and have more conductive property than copper. Such materials may play an expanding role in future development of science and society. Some other materials which are expected to play an important role in future are high temperature superconductors, magnetic materials, biodegradable polymers for packaging, biocompliant solids for surgical implants, etc. Thus, the study of this state becomes more important in the present scenario.
In this Unit, we shall discuss different possible arrangements of particles resulting in several types of structures and explore why different arrangements of structural units lend different properties to solids. We will also learn how these properties get modified due to the structural imperfections or by the presence of impurities in minute amounts.
In Class XI you have learnt that matter can exist in three states namely, solid, liquid and gas. Under a given set of conditions of temperature and pressure, which of these would be the most stable state of a given substance depends upon the net effect of two opposing factors. These are intermolecular forces which tend to keep the molecules (or atoms or ions) closer, and the thermal energy, which tends to keep them apart by making them move faster. At sufficiently low temperature, the thermal energy is low and intermolecular forces bring them so close that they cling to one another and occupy fixed positions. These can still oscillate about their mean positions and the substance exists in solid state. The following are the characteristic properties of the solid state:
1. They have definite mass, volume and shape.
2. Intermolecular distances are short.
3. Intermolecular forces are strong.
4. Their constituent particles (atoms, molecules or ions) have fixed positions and can only oscillate about their mean positions.
5. They are incompressible and rigid.
Solids can be classified as crystalline or amorphous on the basis of the nature of order present in the arrangement of their constituent particles. A crystalline solid usually consists of a large number of small crystals, each of them having a definite characteristic geometrical shape. The arrangement of constituent particles (atoms, molecules or ions) in a crystal is ordered and repetitive in three dimensions. If we observe the pattern in one region of the crystal, we can predict accurately the position of particles in any other region of the crystal however far they may be from the place of observation. Thus, crystal has a long range order which means that there is a regular pattern of arrangement of particles which repeats itself periodically over the entire crystal. Sodium chloride and quartz are typical examples of crystalline solids. Glass, rubber and many plastics do not form crystals when their liquids solidify on cooling. These are called amorphous solids. The term amorphous comes from the Greek word (atoms, molecules or ions) in such a solid has only short range order. In such an arrangement, a regular and periodically repeating pattern is observed over short distances only. Regular patterns are scattered and in between the arrangement is disordered. The structures of quartz (crystalline) and quartz glass (amorphous) are shown in Fig. 1.1 (a) and (b) respectively.
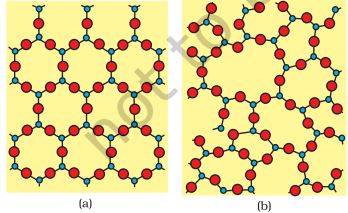
Fig.1.1: Two dimensional structure of quartz and (b) quartz glass
While the two structures are almost identical, yet in the case of amorphous quartz glass there is no long range order. The structure of amorphous solids is similar to that of liquids. Due to the differences in the arrangement of the constituent particles, the two types of solids differ in their properties.
Crystalline solids have a sharp melting point. At a characteristic temperature they melt abruptly and become liquid. On the other hand, amorphous solids soften, melt and start flowing over a range of temperature and can be moulded and blown into various shapes. Amorphous solids have the same structural features as liquids and are conveniently regarded as extremely viscous liquids. They may become crystalline at some temperature. Some glass objects from ancient civilisations are found to become milky in appearance because of some crystallisation. Like liquids, amorphous solids have a tendency to flow, though very slowly. Therefore, sometimes these are called pseudo solids or super cooled liquids.Amorphous solids are isotropic in nature. Their properties such as mechanical strength, refractive index and electrical conductivity, etc., are same in all directions. It is because there is no long range order in them and arrangement of particles is not definite along all the directions. Hence, the overall arrangement becomes equivalent in all directions. Therefore, value of any physical property would be same along any direction.
Crystalline solids are anisotropic in nature, that is, some of their physical properties like electrical resistance or refractive index show different values when measured along different directions in the same crystals. This arises from different arrangement of particles in different directions. This is illustrated in Fig. 1.2.
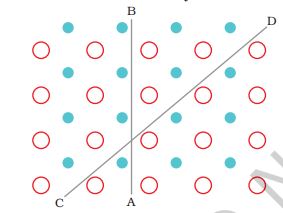
Fig.1.2: Anisotropy in crystals is due to different arrangement of particles along different directions.
This figure shows a simple two- dimensional pattern of arrangement of two kinds of atoms. Mechanical property such as resistance to shearing stress might be quite different in two directions indicated in the figure. Deformation in CD direction displaces row which has two different types of atoms while in AB direction rows made of one type of atoms are displaced. The differences between the crystalline solids and amorphous solids are summarised in Table 1.1.
Table 1.1: Distinction between Crystalline and Amorphous Solids


Besides crystalline and amorphous solids, there are some solids which apparently appear amorphous but have microcrystalline structures. These are called polycrystalline solids. Metals often occur in polycrystalline condition. Individual crystals are randomly oriented so a metallic sample may appear to be isotropic even though a single crystal is anisotropic.
Amorphous solids are useful materials. Glass, rubber and plastics find many applications in our daily lives. Amorphous silicon is one of the best photovoltaic material available for conversion of sunlight into electricity.
Intext Questions
Why are solids rigid?
Why do solids have a definite volume?
Classify the following as amorphous or crystalline solids: Polyurethane, naphthalene, benzoic acid, teflon, potassium nitrate, cellophane, polyvinyl chloride, fibre glass, copper.
Refractive index of a solid is observed to have the same value along all directions. Comment on the nature of this solid. Would it show cleavage property?
Classification of Crystalline Solids
In Section 1.2, we have learnt about amorphous substances and that they have only short range order. However, most of the solid substances are crystalline in nature. For example, all the metallic elements like iron, copper and silver; non-metallic elements like sulphur, phosphorus and iodine and compounds like sodium chloride, zinc sulphide and naphthalene form crystalline solids.
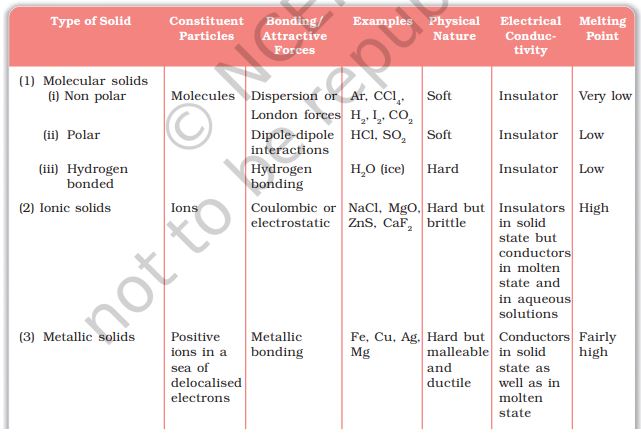
Crystalline solids can be classified in various ways. The method depends on the purpose in hand. Here, we will classify crystalline solids on the basis of nature of intermolecular forces or bonds that hold the constituent particles together. These are — (i) Van der waals forces; (ii)Ionic bonds; (iii) Covalent bonds; and (iv) Metallic bonds. On this basis, crystalline solids are classified into four categories viz., molecular, ionic, metallic and covalent solids. Let us now learn about these categories.
Molecular Solids
Molecules are the constituent particles of molecular solids. These are further sub divided into the following categories:
(1) Non polar Molecular Solids: They comprise either atoms, for example, argon and helium or the molecules formed by non polar covalent
bonds, for example, H2, Cl2 and I2. In these solids, the atoms or molecules are held by weak dispersion forces or London forces about which you have learnt in Class XI. These solids are soft and non-conductors of electricity. They have low melting points and are usually in liquid or gaseous state at room temperature and pressure.(2) Polar Molecular Solids: The molecules of substances like HCl, SO2, etc. are formed by polar covalent bonds. The molecules in such solids are held together by relatively stronger dipole-dipole interactions. These solids are soft and non-conductors of electricity. Their melting points are higher than those of non polar molecular solids yet most of these are gases or liquids under room temperature and pressure. Solid SO2 and solid NH3 are some examples of such solids.
(3)Hydrogen Bonded Molecular Solids: The molecules of such solids contain polar covalent bonds between H and F, O or N atoms. Strong hydrogen bonding binds molecules of such solids like H2O (ice). They are non-conductors of electricity. Generally they are volatile liquids or soft solids under room temperature and pressure.
Ionics Solids
Ions are the constituent particles of ionic solids. Such solids are formed by the three dimensional arrangements of cations and anions bound by strong coulombic (electrostatic) forces. These solids are hard and brittle in nature. They have high melting and boiling points. Since the ions are not free to move about, they are electrical insulators in the solid state. However, in the molten state or when dissolved in water, the ions become free to move about and they conduct electricity.
METALLIC SOLIDS
Metals are orderly collection of positive ions surrounded by and held together by a sea of free electrons. These electrons are mobile and are evenly spread out throughout the crystal. Each metal atom contributes one or more electrons towards this sea of mobile electrons. These free and mobile electrons are responsible for high electrical and thermal conductivity of metals. When an electric field is applied, these electrons flow through the network of positive ions. Similarly, when heat is supplied to one portion of a metal, the thermal energy is uniformly spread throughout by free electrons. Another important characteristic of metals is their lustre and colour in certain cases. This is also due to the presence of free electrons in them. Metals are highly malleable and ductile.
CovaleNT OR Network Solids
A wide variety of crystalline solids of non-metals result from the formation of covalent bonds between adjacent atoms throughout the crystal. They are also called giant molecules. Covalent bonds are strong and directional in nature, therefore atoms are held very strongly at their positions. Such solids are very hard and brittle. They have extremely high melting points and may even decompose before melting. They are insulators and do not conduct electricity. Diamond (Fig. 1.3) and silicon carbide are typical examples of such solids.
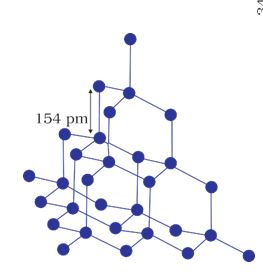
Fig. 1.3: Network structure of diamond
Although Graphite (Fig. 1.4) also belongs to this class of crystals, but it is soft and is a conductor of electricity. Its exceptional properties are due to
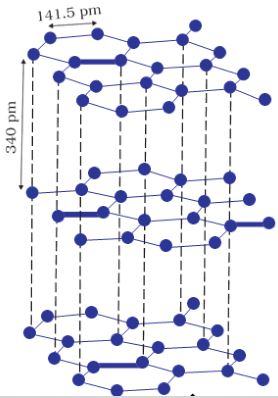
Fig. 1.4: Structure of graphite
its typical structure. Carbon atoms are arranged in different layers and each atom is covalently bonded to three of its neighbouring atoms in the same layer. The fourth valence electron of each atom is present between different layers and is free to move about. These free electrons make graphite a good conductor of electricity. Different layers can slide one over the other. This makes graphite a soft solid and a good solid lubricant.
The different properties of the four types of solids are listed in Table 1.2.
Table 1.2: Different Types of Solids

Classify the following solids in different categories based on the nature of intermolecular forces operating in them:
Potassium sulphate, tin, benzene, urea, ammonia, water, zinc sulphide, graphite, rubidium, argon, silicon carbide.
Solid A is a very hard electrical insulator in solid as well as in molten state and melts at extremely high temperature. What type of solid is it? solids conduct electricity in molten state but not in solid state. Explain. What type of solids are electrical conductors, malleable and ductile?
You must have noticed that when tiles are placed to cover a floor, a repeated pattern is generated. If after setting tiles on floor we mark a point at same location in all the tiles (e.g. Centre of the tile) and see the marked positions only ignoring the tiles, we obtain a set of points. This set of points is the scaffolding on which pattern has been developed by placing tiles. This scaffolding is a space lattice on which two-dimensional pattern has been developed by placing structural units on its set of points (i.e. tile in this case). The structural unit is called basis or motif. When motifs are placed on points in space lattice, a pattern is generated. In crystal structure, motif is a molecule, atom or ion. A space lattice, also called a crystal lattice, is the pattern of points representing the locations of these motifs. In other words, space lattice is an abstract scaffolding for crystal structure. When we place motifs in an identical manner on points of space lattice,
we get crystal structure. Fig. 1.5 shows
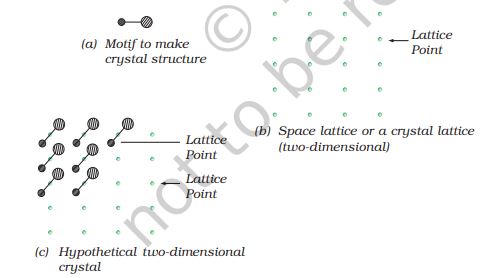
Fig. 1.5: (a) Motif (b) Space lattice (two-dimensional (c) Hypothetical two-dimensional crystal structure
a motif, a two-dimensional lattice and a hypothetical two-dimensional crystal structure obtained by placing motifs in the two-dimensional lattice.
Spacial arrangement of lattice points gives rise to different types of lattices. Fig 1.6 shows arrangement of points in two different lattices.

Fig
In the case of crystalline solids, space lattice is a three-dimensional array of points. The crystal structure is obtained by associating structurral motifs with lattice points. Each repeated basis or motif has same structure and same spacial orientation as other one in a crystal. The environment of each motif is same throughout the crystal except for on surface.
Following are the characteristics of a crystal lattice:
a. Each point in a lattice is called lattice point or lattice site.
b. Each point in a crystal lattice represents one constituent particle which may be an atom, a molecule (group of atoms) or an ion.
c. Lattice points are joined by straight lines to bring out the geometry of the lattice.
We need only a small part of the space lattice of a crystal to spacify crystal completely. This small part is called unit cell. One can choose unit cell in many ways. Normally that cell is chosen which has perpendicular sides of shortest length and one can construct entire crystal by translational displacement of the unit cell in three dimensions. Fig. 1.7 shows

Fig. 1.7: Generating hypothetical two- dimensional crystal structure by shifting square in the direction of arrows.
movement of unit cell of a two-dimensional lattice to construct the entire crystal structure. Also, unit cells have shapes such that these fill the whole lattice without leaving space between cells.
In two dimensions a parallelogram with side of length ‘a’ and ‘b’ and an angle r between these sides is chosen as unit cell. Possible unit cells in two dimensions are shown in Fig. 1.8.
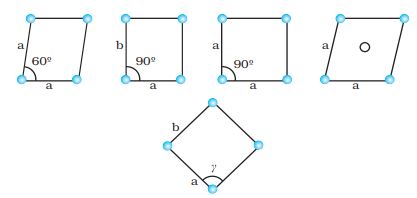
Fig. 1.8: Possible unit cells in two dimensions
A portion of three-dimensional crystal lattice and its unit cell is shown in Fig. 1.9.
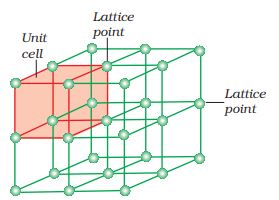
Fig. 1.9: A portion of a three- dimensional cubic space of a crystal lattice and its unit cell
In the three-dimensional crystal structure, unit cell is characterised by:
1. its dimensions along the three edges a, b and c. These edges may or may not be mutually perpendicular.
2. angles between the edges, \(\alpha\) (between b and c), \(\beta\) (between a and c) and \(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGaeq4SdCgaaa!379D! \gamma \) (between a and b). Thus, a unit cell is characterised by six parameters a, b, c, \(\alpha\), \(\beta\) and \(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGaeq4SdCgaaa!379D! \gamma \). These parameters of a typical unit cell are shown in Fig. 1.10.
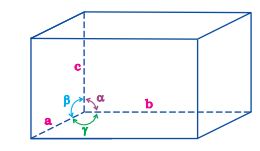
Fig. 1.10: Illustration of parameters of a unit cell
Primitive and Centred Unit Cells
Unit cells can be broadly divided into two categories, primitive and centred unit cells.
(a) Primitive Unit Cells
When constituent particles are present only on the corner positions of a unit cell, it is called as primitive unit cell.
(b) Centred Unit Cells
When a unit cell contains one or more constituent particles present at positions other than corners in addition to those at corners, it is called a centred unit cell. Centred unit cells are of three types:
1. Body-Centred Unit Cells: Such a unit cell contains one constituent particle (atom, molecule or ion) at its body-centre besides the ones that are at its corners.
2. Face-Centred Unit Cells: Such a unit cell contains one constituent particle present at the centre of each face, besides the ones that are at its corners.
3. End-Centred Unit Cells: In such a unit cell, one constituent particle is present at the centre of any two opposite faces besides the ones present at its corners.
Inspection of a wide variety of crystals leads to the conclusion that all can be regarded as conforming to one of the seven regular figures. These basic regular figures are called seven crystal systems. To which system a given crystal belongs to is determined by measuring the angles between its faces and deciding how many axis are needed to define the principal features of its shape. Fig. 1.11 shows seven crystal systems.
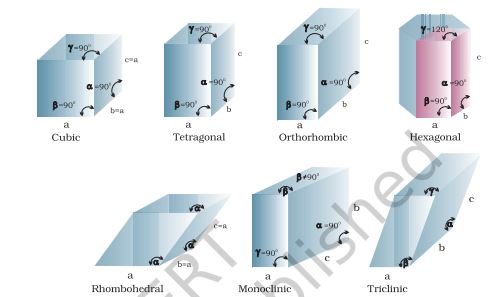
Fig. 1.11. Seven crystal systems
A French mathematician, Bravais, showed that there are only 14 possible three-dimensional lattices. These are called Bravais lattices. Unit cells of these lattices are shown in the following box. The characteristics of their primitive unit cells along with the centred unit cells that they can form have been listed in Table 1.3.
Table 1.3: Seven Primitive Unit Cells and their Possible Variations as Centred Unit Cells

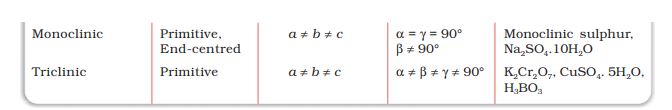
NUMBER OF ATOMS IN A UNIT CELL
We know that any crystal lattice is made up of a very large number of unit cells and every lattice point is occupied by one constituent particle (atom, molecule or ion). Let us now work out what portion of each particle belongs to a particular unit cell.
We shall consider three types of cubic unit cells and for simplicity assume that the constituent particle is an atom.
PRIMITIVE CUBIC UNIT CELL
Primitive cubic unit cell has atoms only at its corner. Each atom at a corner is shared between eight adjacent unit cells as shown in Fig. 1.12, four unit cells in the same layer and four unit cells of the

Fig. 1.12: In a simple cubic unit cell, each corner atom is shared between 8 unit cells.
upper (or lower) layer. Therefore, only \(\frac{1}{8}\) th of an atom (or molecule or ion) actually belongs to a particular unit cell. In Fig. 1.13, a primitive cubic unit cell has been depicted in three different ways.
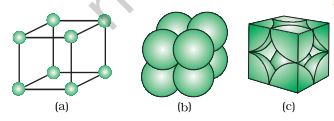
Fig. 1.13: A primitive cubic unit cell (a) open structure (b) space-filling structure (c) actual portions of atoms belonging to one unit cell.
Each small sphere in Fig. 1.13(a) represents only the centre of the particle occupying that position and not its actual size. Such structures are called open structures. The arrangement of particles is easier to follow in open structures. Fig. 1.13 (b) depicts space-filling representation of the unit cell with actual particle size and Fig. 1.13 (c) shows the actual portions of different atoms present in a cubic unit cell.
In all, since each cubic unit cell has 8 atoms on its corners, the total number of atoms in one unit cell is \(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGaaGioaiabgE % na0oaalaaabaGaaGymaaqaaiaaiIdaaaGaeyypa0JaaGymaaaa!3C1D! 8 \times \frac{1}{8} = 1\) atom.
BODY CENTRED CUBIC UNIT
A body-centred cubic (bcc) unit cell has an atom at each of its corners and also one atom at its body centre. Fig. 1.14 depicts (a) open structure (b) space filling model and (c) the unit cell with portions of atoms actually belonging to it. It can be seen that the atom at the
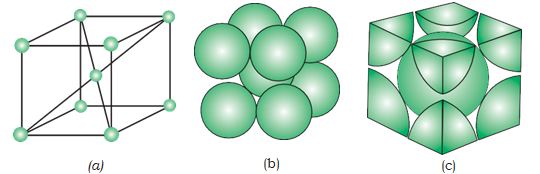
Fig. 1.14: A body-centred cubic unit cell (a) open structure (b) space- filling structure (c) actual portions of atoms belonging to one unit cell.
body centre wholly belongs to the unit cell in which it is present. Thus in a body-centered cubic (bcc) unit cell:
(1) 8 corners ×\(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaWaaSaaaeaaca % aIXaaabaGaaGioaaaaaaa!3783! \frac{1}{8}\) per corner atom \(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGaaGioaiabgE % na0oaalaaabaGaaGymaaqaaiaaiIdaaaGaeyypa0JaaGymaaaa!3C1D! 8 \times \frac{1}{8} = 1\) atom
(2) 1 body centre atom = 1 × 1 = 1 atom
\(\therefore\)Total number of atoms per unit cell = 2 atoms
A face-centred cubic (fcc) unit cell contains atoms at all the corners and at the centre of all the faces of the cube. It can be seen in Fig. 1.15

Fig. 1.15: An atom at face centre of unit cell is shared between 2 unit cells
that each atom located at the face-centre is shared between two adjacent unit cells and only \(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaWaaSaaaeaaca % aIXaaabaGaaGOmaaaaaaa!377D! \frac{1}{2}\) of each atom belongs to a unit cell. Fig. 1.16 depicts
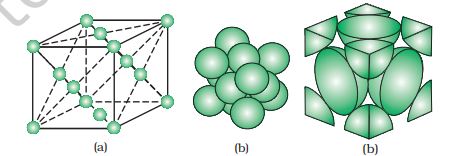
Fig 1.16: A face-centred cubic unit cell (a) open structure (b) space filling structure (c) actual portions of atoms belonging to one unit cell.
(a) open structure (b) space-filling model and (c) the unit cell with portions of atoms actually belonging to it. Thus, in a face-centred cubic (fcc) unit cell:
(1) 8 corners atoms \(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGaey41aq7aaS % aaaeaacaaIXaaabaGaaGioaaaaaaa!399A! \times \frac{1}{8}\) atom per unit cell \(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGaaGioaiabgE % na0oaalaaabaGaaGymaaqaaiaaiIdaaaGaeyypa0JaaGymaaaa!3C1D!8 \times \frac{1}{8} = 1\) atom
(2) 6 face-centred atoms \(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGaey41aq7aaS % aaaeaacaaIXaaabaGaaGOmaaaaaaa!3994! \times \frac{1}{2}\) atom per unit cell \(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGaaGOnaiabgE % na0oaalaaabaGaaGymaaqaaiaaikdaaaGaeyypa0JaaG4maaaa!3C17! 6 \times \frac{1}{2} = 3\) atoms
\(\therefore\)Total number of atoms per unit cell = 4 atoms
INTEXT QUESTION
Give the significance of a ‘lattice point’.
Name the parameters that characterise a unit cell. Distinguish between
1. Hexagonal and monoclinic unit cells
2. Face-centred and end-centred unit cells.
Explain how much portion of an atom located at (i) corner and (ii) body- centre of a cubic unit cell is part of its neighbouring unit cell.
CLOSED PACKED STRUCTURE
In solids, the constituent particles are close-packed, leaving the minimum vacant space. Let us consider the constituent particles as identical hard spheres and build up the three-dimensional structure in three steps.
(a) Close Packing in One Dimension
There is only one way of arranging spheres in a one-dimensional close packed structure, that is to arrange them in a row and touching each other (Fig. 1.17).

Fig. 1.17: Close packing of spheres in one dimension
In this arrangement, each sphere is in contact with two of its neighbours. The number of nearest neighbours of a particle is called it coordination number Thus, in one dimensional close packed arrangement, the coordination number is 2.
(b) Close Packing in Two Dimensions
Two dimensional close packed structure can be generated by stacking (placing) the rows of close packed spheres. This can be done in two different ways.
(1) The second row may be placed in contact with the first one such that the spheres of the second row are exactly above those of the first row. The spheres of the two rows are aligned horizontally as well as vertically. If we call the first row as ‘A’ type row, the second row being exactly the same as the first one, is also of ‘A’ type. Similarly, we may place more rows to obtain AAA type of arrangement as shown in Fig. 1.18 (a).
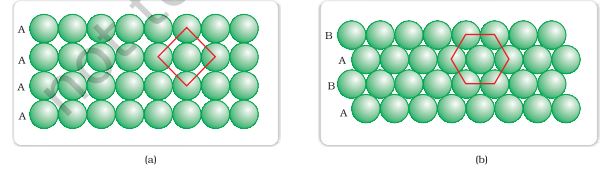
Fig. 1.18: (a) Square close packing (b) hexagonal close packing of spheres in two dimensions
In this arrangement, each sphere is in contact with four of its neighbours. Thus, the two dimensional coordination number is 4. Also, if the centres of these 4 immediate neighbouring spheres are joined, a square is formed. Hence this packing is called square close packing in two dimensions.(2) The second row may be placed above the first one in a staggered manner such that its spheres fit in the depressions of the first row. If the arrangement of spheres in the first row is called ‘A’ type, the one in the second row is different and may be called ‘B’ type. When the third row is placed adjacent to the second in staggered manner, its spheres are aligned with those of the first layer. Hence this layer is also of ‘A’ type. The spheres of similarly placed fourth row will be aligned with those of the second row (‘B’ type). Hence this arrangement is of ABAB type. In this arrangement there is less free space and this packing is more efficient than the square close packing. Each sphere is in contact with six of its neighbours and the two dimensional coordination number is 6. The centres of these six spheres are at the corners of a regular hexagon (Fig. 1.18 b) hence this packing is called two dimensional hexagonal close- packing. It can be seen in Figure 1.18 (b) that in this layer there are some voids (empty spaces). These are triangular in shape. The triangular voids are of two different types. In one row, the apex of the triangles are pointing upwards and in the next layer downwards.
(c) Close Packing in Three Dimensions
All real structures are three dimensional structures. They can be obtained by stacking two dimensional layers one above the other. In the last Section, we discussed close packing in two dimensions which can be of two types; square close-packed and hexagonal close-packed. Let us see what types of three dimensional close packing can be obtained from these.
(1) Three-dimensional close packing forms two-dimensional square close-packed layers: While placing the second square close-packed layer above the first we follow the same rule that was followed when one row was placed adjacent to the other. The second layer is placed over the first layer such that the spheres of the upper layer are exactly above those of the first layer. In this arrangement spheres of both the layers are perfectly aligned horizontally as well as vertically as shown in Fig. 1.19.
Fig. 1.19: Simple cubic lattice formed by A A A arrangement
Similarly, we may place more layers one above the other. If the arrangement of spheres in the first layer is called ‘A’ type, all the layers have the same arrangement. Thus this lattice has AAA.... type pattern. The lattice thus generated is the simple cubic lattice, and its unit cell is the primitive cubic unit cell (See Fig. 1.19).
(2) Three dimensional close packing from two dimensional hexagonal close packed layers: Three dimensional close packed structure can be generated by placing layers one over the other.
(2) Three dimensional close packing from two dimensional hexagonal close packed layers: Three dimensional close packed structure can be generated by placing layers one over the other.
(a) ) Placing second layer over the first layer
Let us take a two dimensional hexagonal close packed layer ‘A’ and place a similar layer above it such that the spheres of the second layer are placed in the depressions of the first layer. Since the spheres of the two layers are aligned differently, let us call the second layer as B. It can be observed from Fig. 1.20
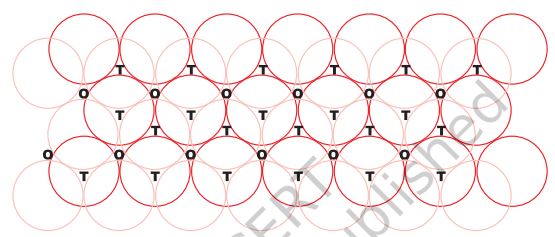
Fig. 1.20: A stack of two layers of close packed spheres and voids generated in them. T = Tetrahedral void; O = Octahedral void
that all the triangular voids of the first layer are not covered by the spheres of the second layer. This to different arrangements. Wherever a sphere of the second layer is above the void of the first layer (or vice versa) a tetrahedral void is formed. These voids are called tetrahedral voids because a tetrahedron is formed when the centres of these four spheres are joined. They have been marked as ‘T’ in Fig. 1.20. One such void has been shown separately in Fig. 1.21.
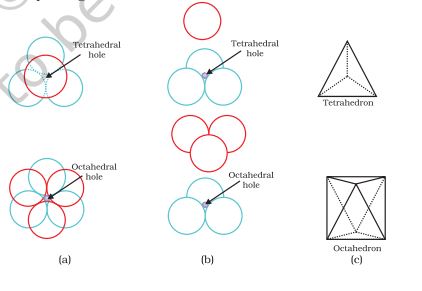
Fig 1.21 Tetrahedral and octahedral voids
(a) top view
(b) exploded side view and
(c) geometrical shape of the void.
At other places, the triangular voids in the second layer are above the triangular voids in the first layer, and the triangular shapes of these do not overlap. One of them has the apex of the triangle pointing upwards and the other downwards. These voids have been marked as ‘O’ in Fig. 1.20. Such voids are surrounded by six spheres and are called octahedral voids. One such void has been shown separately in Fig. 1.21. The number of these two types of voids depend upon the number of close packed spheres.
Let the number of close packed spheres be N, then:
The number of octahedral voids generated = N
The number of tetrahedral voids generated = 2N
(b) Placing third layer over the second layer
When third layer is placed over the second, there are two possibilities.
1. Covering Tetrahedral Voids: Tetrahedral voids of the second layer may be covered by the spheres of the third layer. In this case, the spheres of the third layer are exactly aligned with those of the first layer. Thus, the pattern of spheres is repeated in alternate layers. This pattern is often written as ABAB pattern. This structure is called hexagonal close packed (hcp) structure (Fig. 1.22). This sort of arrangement of atoms is found in many metals like magnesium and zinc.
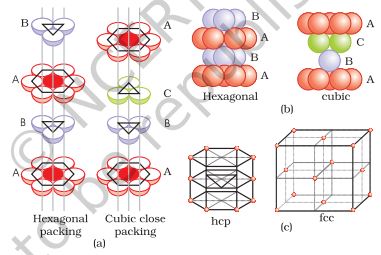
Fig. 1.22
a. Hexagonal cubic close-packing exploded view showing stacking of layers of spheres
b.four layers stacked in each case and (c) geometry of packing.
(2) Covering Octahedral Voids: The third layer may be placed above the second layer in a manner such that its spheres cover the octahedral voids. When placed in this manner, the spheres of the third layer are not aligned with those of either the first or the second layer. This arrangement is called ‘C’ type. Only when fourth layer is placed, its spheres are aligned with those of the first layer as shown in Figs. 1.22 and 1.23. This pattern of layers is often written as ABCABC......................................................................... This structure is called cubic close packed (ccp) or face-centred cubic (fcc) structure. Metals such as copper and silver crystallise in this structure.
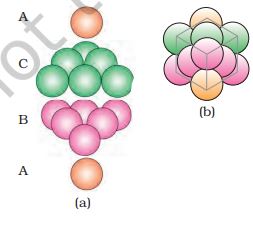
Figure 1.23.
(a) ABCABC... arrangement of layers when octahedral void is covered (b) fragment of structure formed by this arrangement resulting in cubic
closed packed (ccp) or face centred cubic (fcc) structure
FORMULA OF A Compound and Number of Voids Filled
Earlier in the section, we have learnt that when particles are close- packed resulting in either ccp or hcp structure, two types of voids are generated. While the number of octahedral voids present in a lattice is equal to the number of close packed particles, the number of tetrahedral voids generated is twice this number. In ionic solids, the bigger ions (usually anions) form the close packed structure and the smaller ions (usually cations) occupy the voids. If the latter ion is small enough then tetrahedral voids are occupied, if bigger, then octahedral voids. All octahedral or tetrahedral voids are not occupied. In a given compound, the fraction of octahedral or tetrahedral voids that are occupied, depends upon the chemical formula of the compound, as can be seen from the following examples.
EXAMPLE 1
A compound is formed by two elements X and Y. Atoms of the element Y (as anions) make ccp and those of the element X (as cations) occupy all the octahedral voids. What is the formula of the compound?
SOLUTION
The ccp lattice is formed by the element Y. The number of octahedral voids generated would be equal to the number of atoms of Y present in it. Since all the octahedral voids are occupied by the atoms of X, their number would also be equal to that of the element Y. Thus, the atoms of elements X and Y are present in equal numbers or 1:1 ratio. Therefore, the formula of the compound is XY.
EXAMPLE 2
Atoms of element B form hcp lattice and those of the element A occupy 2/3rd of tetrahedral voids. What is the formula of the compound formed by the elements A and B?
SOLUTION
The number of tetrahedral voids formed is equal to twice the number of atoms of element B and only 2/3rd of these are occupied by the atoms of element A. Hence the ratio of the number of atoms of A and B is 2 × (2/3):1 or 4:3 and the formula of the compound is A4B3.
Locating Tetrahedral and Octahedral Voids
We know that close packed structures have both tetrahedral and octahedral voids. Let us take ccp (or fcc) structure and locate these voids in it.
a. Locating Tetrahedral Voids
Let us consider a unit cell of ccp or fcc lattice [Fig. 1(a)]. The unit cell is divided into eight small cubes.
Each small cube has atoms at alternate corners [Fig. 1(a)]. In all, each small cube has 4 atoms. When joined to each other, they make a regular tetrahedron. Thus, there is one tetrahedral void in each small cube and eight tetrahedral voids in total. Each of the eight small cubes have one void in one unit cell of ccp structure. We know that ccp structure has 4 atoms per unit cell. Thus, the number of tetrahedral voids is twice the number of atoms.
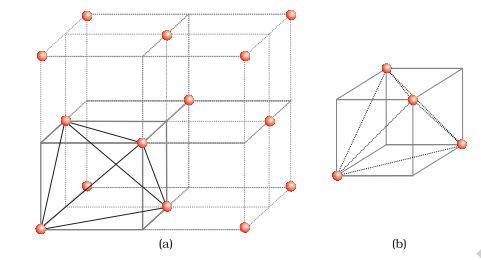
Fig. 1: (a) Eight tetrahedral voids per unit cell of ccp structure
(b) one tetrahedral void showing the geometry.
(b) Locating Octahedral Voids
Let us again consider a unit cell of ccp or fcc lattice [Fig. 2(a)]. The body centre of the cube, C is not occupied but it is surrounded by six atoms on face centres. If these face centres are joined, an octahedron is generated. Thus, this unit cell has one octahedral void at the body centre of the cube.
Besides the body centre, there is one octahedral void at the centre of each of the 12 edges [Fig. 2(b)]. It is surrounded by six atoms, four belonging to the same unit cell (2 on the corners and 2 on face centre) and two belonging to two adjacent unit cells. Since each edge of the cube is shared between four adjacent unit cells, so is the octahedral void located on it. Only \(\frac{1}{4}\)th of each void belongs to a particular unit cell.
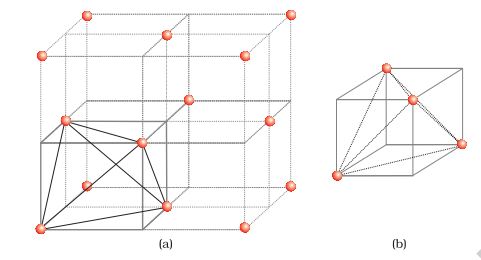
Fig. 2: Location of octahedral voids per unit cell of ccp or fcc lattice (a) at the body centre of the cube and (b) at the centre of each edge (only one such void is shown).
Thus in cubic close packed structure:
Octahedral void at the body-centre of the cube = 1
12 octahedral voids located at each edge and shared between four unit cells
\(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGaeyypa0JaaG % ymaiaaikdacqGHxdaTdaWcaaqaaiaaigdaaeaacaaI0aaaaiabg2da % 9iaaiodaaaa!3DD6! = 12 \times \frac{1}{4} = 3\)
\(\therefore\)Total number of octahedral voids = 4
We know that in ccp structure, each unit cell has 4 atoms. Thus, the number of octahedral voids is equal to this number.
PACKING EfficienCY
In whatever way the constituent particles (atoms, molecules or ions) are packed, there is always some free space in the form of voids. Packing efficiency is the percentage of total space filled by the particles. Let us calculate the packing efficiency in different types of structures.
PACKING EfficienCY hcp and ccp Structures
Both types of close packing (hcp and ccp) are equally efficient. Let us calculate the efficiency of packing in ccp structure. In Fig. 1.24 let the unit cell edge length be ‘a’ and face diagonal AC = b.
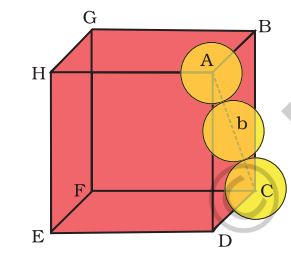
Fig. 1.24: Cubic close packing other sides are not provided with spheres for sake of clarity
If \(\Delta\)ABC
\(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGceaqabeaacaWGbb % Gaam4qamaaCaaaleqabaGaaGOmaaaakiabg2da9iaadkgadaahaaWc % beqaaiaaikdaaaGccqGH9aqpcaWGcbGaam4qamaaCaaaleqabaGaaG % OmaaaakiabgUcaRiaadgeacaWGcbWaaWbaaSqabeaacaaIYaaaaaGc % baGaeyypa0JaamyyamaaCaaaleqabaGaaGOmaaaakiabgUcaRiaadg % gadaahaaWcbeqaaiaaikdaaaGccqGH9aqpcaaIYaGaamyyamaaCaaa % leqabaGaaGOmaaaakiaaykW7caaMc8UaaGPaVlaad+gacaWGYbGaaG % PaVlaaykW7caaMc8UaaGPaVdqaaiaadkgacqGH9aqpcaaI0aGaamOC % aiabg2da9maakaaabaGaaGOmaaWcbeaakiaadggaaaaa!5EA5! \begin{array}{l} A{C^2} = {b^2} = B{C^2} + A{B^2}\\ = {a^2} + {a^2} = 2{a^2}\,\,\,or\,\,\,\,\\ b = 4r = \sqrt 2 a \end{array}\)
If r is the radius of the sphere, we find
\(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGceaqabeaacaWGIb % Gaeyypa0JaaGinaiaadkhacqGH9aqpdaGcaaqaaiaaikdaaSqabaGc % caWGHbaabaGaam4BaiaadkhacaaMc8UaaGPaVlaadggacqGH9aqpda % WcaaqaaiaaisdacaWGYbaabaWaaOaaaeaacaaIYaaaleqaaaaakiab % g2da9iaaikdadaGcaaqaaiaaikdaaSqabaGccaWGYbaaaaa!4999! \begin{array}{l} b = 4r = \sqrt 2 a\\ or\,\,a = \frac{{4r}}{{\sqrt 2 }} = 2\sqrt 2 r \end{array}\)
(we can also write, r = \(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGaamOCaiabg2 % da9maalaaabaGaamyyaaqaaiaaikdadaGcaaqaaiaaikdaaSqabaaa % aaaa!3A7C! r = \frac{a}{{2\sqrt 2 }}\))
We know, that each unit cell in ccp structure, has effectively 4 spheres. Total volume of four spheres is equal to \(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGaaGinaiabgE % na0oaabmaabaGaaGinaiaac+cacaaIZaaacaGLOaGaayzkaaGaeqiW % daNaamOCamaaCaaaleqabaGaaG4maaaaaaa!4020! 4 \times \left( {4/3} \right)\pi {r^3}\) and volume of the cube is a3 or \(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaWaaeWaaeaaca % aIYaWaaOaaaeaacaaIYaaaleqaaOGaamOCaaGaayjkaiaawMcaamaa % CaaaleqabaGaaG4maaaaaaa!3AFD! {\left( {2\sqrt 2 r} \right)^3}\)
Therefore,
\(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGceaqabeaacaWGqb % GaamyyaiaadogacaWGRbGaamyAaiaad6gacaWGNbGaaGPaVlaaykW7 % caWGLbGaamOzaiaadAgacaWGPbGaam4yaiaadMgacaWGLbGaamOBai % aadogacaWG5bGaeyypa0ZaaSaaaeaacaWGwbGaam4BaiaadYgacaWG % 1bGaamyBaiaadwgacaaMc8UaaGPaVlaad+gacaWGJbGaam4yaiaadw % hacaWGWbGaamyAaiaadwgacaWGKbGaaGPaVlaaykW7caWGIbGaamyE % aiaaykW7caaMc8UaamOzaiaad+gacaWG1bGaamOCaiaaykW7caaMc8 % Uaam4CaiaadchacaWGObGaamyzaiaadkhacaWGLbGaam4CaiaaykW7 % caaMc8UaamyAaiaad6gacaaMc8UaamiDaiaadIgacaWGLbGaaGPaVl % aaykW7caWG1bGaamOBaiaadMgacaWG0bGaaGPaVlaaykW7caWGJbGa % amyzaiaadYgacaWGSbGaey41aqRaaGymaiaaicdacaaIWaaabaGaam % ivaiaad+gacaWG0bGaamyyaiaadYgacaaMc8UaaGPaVlaadAhacaWG % VbGaamiBaiaadwhacaWGTbGaamyzaiaaykW7caaMc8Uaam4BaiaadA % gacaaMc8UaaGPaVlaadshacaWGObGaamyzaiaaykW7caaMc8UaamyD % aiaad6gacaWGPbGaamiDaiaaykW7caaMc8Uaam4yaiaadwgacaWGSb % GaamiBaaaacaGGLaaabaGaaGPaVlaaykW7caaMc8UaaGPaVlaaykW7 % caaMc8UaaGPaVlaaykW7caaMc8UaaGPaVlaaykW7caaMc8UaaGPaVl % aaykW7caaMc8UaaGPaVlaaykW7caaMc8UaaGPaVlaaykW7caaMc8Ua % aGPaVlaaykW7caaMc8UaaGPaVlaaykW7caaMc8UaaGPaVlaaykW7ca % aMc8UaaGPaVlaaykW7caaMc8UaaGPaVlaaykW7caaMc8UaaGPaVlaa % ykW7caaMc8UaaGPaVlaaykW7cqGH9aqpdaWcaaqaaiaaisdacqGHxd % aTdaqadaqaaiaaisdacaGGVaGaaG4maaGaayjkaiaawMcaaiabec8a % WjaadkhadaahaaWcbeqaaiaaiodaaaGccqGHxdaTcaaIXaGaaGimai % aaicdaaeaadaqadaqaaiaaikdadaGcaaqaaiaaikdaaSqabaGccaWG % YbaacaGLOaGaayzkaaWaaWbaaSqabeaacaaIZaaaaaaakiaacwcaae % aacaaMc8UaaGPaVlaaykW7caaMc8UaaGPaVlaaykW7caaMc8UaaGPa % VlaaykW7caaMc8UaaGPaVlaaykW7caaMc8UaaGPaVlaaykW7caaMc8 % UaaGPaVlaaykW7caaMc8UaaGPaVlaaykW7caaMc8UaaGPaVlaaykW7 % caaMc8UaaGPaVlaaykW7caaMc8UaaGPaVlaaykW7caaMc8UaaGPaVl % aaykW7caaMc8UaaGPaVlaaykW7caaMc8UaaGPaVlaaykW7caaMc8Ua % aGPaVlabg2da9maalaaabaWaaeWaaeaacaaIXaGaaGOnaiaac+caca % aIZaaacaGLOaGaayzkaaGaeqiWdaNaamOCamaaCaaaleqabaGaaG4m % aaaakiabgEna0kaaigdacaaIWaGaaGimaaqaaiaaigdacaaI2aWaaO % aaaeaacaaIYaaaleqaaOGaamOCamaaCaaaleqabaGaaG4maaaaaaGc % caGGLaaaaaa!57AA! \begin{array}{l} Packing\,\,efficiency = \frac{{Volume\,\,occupied\,\,by\,\,four\,\,spheres\,\,in\,the\,\,unit\,\,cell \times 100}}{{Total\,\,volume\,\,of\,\,the\,\,unit\,\,cell}}\% \\ \,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\, = \frac{{4 \times \left( {4/3} \right)\pi {r^3} \times 100}}{{{{\left( {2\sqrt 2 r} \right)}^3}}}\% \\ \,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\, = \frac{{\left( {16/3} \right)\pi {r^3} \times 100}}{{16\sqrt 2 {r^3}}}\% \end{array}\)
Efficiency of Packing in Body-Centred Cubic Structures
From Fig. 1.25, it is clear that the atom at the centre will be in touch with the other two atoms diagonally arranged.

Fig. 1.25: Body-centred cubic unit cell (sphere along the body diagonal are shown with solid boundaries).
If \(\Delta\)EFD
\(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGceaqabeaacaWGIb % WaaWbaaSqabeaacaaIYaaaaOGaeyypa0JaamyyamaaCaaaleqabaGa % aGOmaaaakiabgUcaRiaadggadaahaaWcbeqaaiaaikdaaaGccqGH9a % qpcaaIYaGaamyyamaaCaaaleqabaGaaGOmaaaaaOqaaiaadkgacqGH % 9aqpdaGcaaqaaiaaikdaaSqabaGccaWGHbaabaGaamOtaiaad+gaca % WG3bGaaGPaVlaaykW7caWGPbGaamOBaiaaykW7caaMc8UaeyiLdqKa % amyqaiaadAeacaWGebaabaGaam4yamaaCaaaleqabaGaaGOmaaaaki % abg2da9iaadggadaahaaWcbeqaaiaaikdaaaGccqGHRaWkcaWGIbWa % aWbaaSqabeaacaaIYaaaaOGaeyypa0JaamyyamaaCaaaleqabaGaaG % OmaaaakiabgUcaRiaaikdacaWGHbWaaWbaaSqabeaacaaIYaaaaOGa % eyypa0JaaG4maiaadggadaahaaWcbeqaaiaaikdaaaaakeaacaWGJb % Gaeyypa0ZaaOaaaeaacaaIZaaaleqaaOGaamyyaaaaaa!6872! \begin{array}{l} {b^2} = {a^2} + {a^2} = 2{a^2}\\ b = \sqrt 2 a\\ Now\,\,in\,\,\Delta AFD\\ {c^2} = {a^2} + {b^2} = {a^2} + 2{a^2} = 3{a^2}\\ c = \sqrt 3 a \end{array}\)
The length of the body diagonal c is equal to 4r, where r is the radius of the sphere (atom), as all the three spheres along the diagonal touch each other.
Therefore,
\(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGceaqabeaadaGcaa % qaaiaaiodaaSqabaGccaWGHbGaeyypa0JaaGinaiaadkhaaeaacaWG % HbGaeyypa0ZaaSaaaeaacaaI0aGaamOCaaqaamaakaaabaGaaG4maa % Wcbeaaaaaaaaa!3F09! \begin{array}{l} \sqrt 3 a = 4r\\ a = \frac{{4r}}{{\sqrt 3 }} \end{array}\)
Also we can write, \(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGaamOCaiabg2 % da9maalaaabaWaaOaaaeaacaaIZaaaleqaaaGcbaGaaGinaaaacaWG % Hbaaaa!3A89! r = \frac{{\sqrt 3 }}{4}a\)
In this type of structure, total number of atoms is 2 and their volume is \(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGaaGOmaiabgE % na0oaabmaabaWaaSaaaeaacaaI0aaabaGaaG4maaaaaiaawIcacaGL % PaaacqaHapaCcaWGYbWaaWbaaSqabeaacaaIZaaaaaaa!3F7B! 2 \times \left( {\frac{4}{3}} \right)\pi {r^3}\).
Volume of the cube, a3 will be equal to \(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaWaaeWaaeaada % WcaaqaaiaaisdaaeaadaGcaaqaaiaaiodaaSqabaaaaOGaamOCaaGa % ayjkaiaawMcaamaaCaaaleqabaGaaG4maaaakiaaykW7caaMc8Uaam % 4BaiaadkhacaaMc8UaaGPaVlaadggadaahaaWcbeqaaiaaiodaaaGc % cqGH9aqpdaqadaqaamaalaaabaGaaGinaaqaamaakaaabaGaaG4maa % WcbeaaaaGccaWGYbaacaGLOaGaayzkaaWaaWbaaSqabeaacaaIZaaa % aaaa!4B2B! {\left( {\frac{4}{{\sqrt 3 }}r} \right)^3}\,\,or\,\,{a^3} = {\left( {\frac{4}{{\sqrt 3 }}r} \right)^3}\)
\(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGceaqabeaacaWGqb % GaamyyaiaadogacaWGRbGaamyAaiaad6gacaWGNbGaaGPaVlaaykW7 % caWGLbGaamOzaiaadAgacaWGPbGaam4yaiaadMgacaWGLbGaamOBai % aadogacaWG5bGaeyypa0ZaaSaaaeaacaWGwbGaam4BaiaadYgacaWG % 1bGaamyBaiaadwgacaaMc8UaaGPaVlaad+gacaWGJbGaam4yaiaadw % hacaWGWbGaamyAaiaadwgacaWGKbGaaGPaVlaaykW7caWGIbGaamyE % aiaaykW7caaMc8UaamOzaiaad+gacaWG1bGaamOCaiaaykW7caaMc8 % Uaam4CaiaadchacaWGObGaamyzaiaadkhacaWGLbGaam4CaiaaykW7 % caaMc8UaamyAaiaad6gacaaMc8UaamiDaiaadIgacaWGLbGaaGPaVl % aaykW7caWG1bGaamOBaiaadMgacaWG0bGaaGPaVlaaykW7caWGJbGa % amyzaiaadYgacaWGSbGaey41aqRaaGymaiaaicdacaaIWaaabaGaam % ivaiaad+gacaWG0bGaamyyaiaadYgacaaMc8UaaGPaVlaadAhacaWG % VbGaamiBaiaadwhacaWGTbGaamyzaiaaykW7caaMc8Uaam4BaiaadA % gacaaMc8UaaGPaVlaadshacaWGObGaamyzaiaaykW7caaMc8UaamyD % aiaad6gacaWGPbGaamiDaiaaykW7caaMc8Uaam4yaiaadwgacaWGSb % GaamiBaaaacaGGLaaabaGaaGPaVlaaykW7caaMc8UaaGPaVlaaykW7 % caaMc8UaaGPaVlaaykW7caaMc8UaaGPaVlaaykW7caaMc8UaaGPaVl % aaykW7caaMc8UaaGPaVlaaykW7caaMc8UaaGPaVlaaykW7caaMc8Ua % aGPaVlaaykW7caaMc8UaaGPaVlaaykW7caaMc8UaaGPaVlaaykW7ca % aMc8UaaGPaVlaaykW7caaMc8UaaGPaVlaaykW7caaMc8UaaGPaVlaa % ykW7caaMc8UaaGPaVlaaykW7caaMc8Uaeyypa0ZaaSaaaeaacaaIYa % Gaey41aq7aaeWaaeaacaaI0aGaai4laiaaisdaaiaawIcacaGLPaaa % cqaHapaCcaWGYbWaaWbaaSqabeaacaaIZaaaaOGaey41aqRaaGymai % aaicdacaaIWaaabaWaamWaaeaadaqadaqaaiaaisdacaGGVaWaaOaa % aeaacaaIZaaaleqaaaGccaGLOaGaayzkaaGaamOCaaGaay5waiaaw2 % faamaaCaaaleqabaGaaG4maaaaaaGccaGGLaaabaGaaGPaVlaaykW7 % caaMc8UaaGPaVlaaykW7caaMc8UaaGPaVlaaykW7caaMc8UaaGPaVl % aaykW7caaMc8UaaGPaVlaaykW7caaMc8UaaGPaVlaaykW7caaMc8Ua % aGPaVlaaykW7caaMc8UaaGPaVlaaykW7caaMc8UaaGPaVlaaykW7ca % aMc8UaaGPaVlaaykW7caaMc8UaaGPaVlaaykW7caaMc8UaaGPaVlaa % ykW7caaMc8UaaGPaVlaaykW7caaMc8UaaGPaVlaaykW7cqGH9aqpda % WcaaqaamaabmaabaGaaGioaiaac+cacaaIZaaacaGLOaGaayzkaaGa % eqiWdaNaamOCamaaCaaaleqabaGaaG4maaaakiabgEna0kaaigdaca % aIWaGaaGimaaqaaiaaiAdacaaI0aGaai4lamaabmaabaGaaG4mamaa % kaaabaGaaG4maaWcbeaaaOGaayjkaiaawMcaaiaadkhadaahaaWcbe % qaaiaaiodaaaaaaOGaaiyjaiabg2da9iaaiAdacaaI4aGaaiyjaaaa % aa!6151! \begin{array}{l} Packing\,\,efficiency = \frac{{Volume\,\,occupied\,\,by\,\,four\,\,spheres\,\,in\,the\,\,unit\,\,cell \times 100}}{{Total\,\,volume\,\,of\,\,the\,\,unit\,\,cell}}\% \\ \,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\, = \frac{{2 \times \left( {4/4} \right)\pi {r^3} \times 100}}{{{{\left[ {\left( {4/\sqrt 3 } \right)r} \right]}^3}}}\% \\ \,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\, = \frac{{\left( {8/3} \right)\pi {r^3} \times 100}}{{64/\left( {3\sqrt 3 } \right){r^3}}}\% = 68\% \end{array}\)
PACKING Efficiency in Simple Cubic Lattice
In a simple cubic lattice the atoms are located only on the corners of the cube. The particles touch each other along the edge (Fig. 1.26).
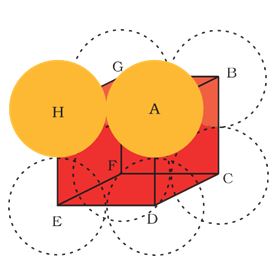
Fig. 1.26
Simple cubic unit cell. The spheres are in contact with each other along the edge of the cube.
Thus, the edge length or side of the cube ‘a’, and the radius of each particle, r are related as
a = 2r
The volume of the cubic unit cell = a3 = (2r)3 = 8r3
Since a simple cubic unit cell contains only 1 atom
The volume of the occupied space = \(\frac{4}{3}\pi\)r3\(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGceaqabeaacqGH0i % cxcaWGqbGaamyyaiaadogacaWGRbGaamyAaiaad6gacaWGNbGaaGPa % VlaaykW7caWGLbGaamOzaiaadAgacaWGPbGaam4yaiaadMgacaWGLb % GaamOBaiaadogacaWG5bGaeyypa0ZaaSaaaeaacaWGwbGaam4Baiaa % dYgacaWG1bGaamyBaiaadwgacaaMc8UaaGPaVlaad+gacaWGJbGaam % 4yaiaadwhacaWGWbGaamyAaiaadwgacaWGKbGaaGPaVlaaykW7caWG % IbGaamyEaiaaykW7caaMc8UaamOzaiaad+gacaWG1bGaamOCaiaayk % W7caaMc8Uaam4CaiaadchacaWGObGaamyzaiaadkhacaWGLbGaam4C % aiaaykW7caaMc8UaamyAaiaad6gacaaMc8UaamiDaiaadIgacaWGLb % GaaGPaVlaaykW7caWG1bGaamOBaiaadMgacaWG0bGaaGPaVlaaykW7 % caWGJbGaamyzaiaadYgacaWGSbGaey41aqRaaGymaiaaicdacaaIWa % aabaGaamivaiaad+gacaWG0bGaamyyaiaadYgacaaMc8UaaGPaVlaa % dAhacaWGVbGaamiBaiaadwhacaWGTbGaamyzaiaaykW7caaMc8Uaam % 4BaiaadAgacaaMc8UaaGPaVlaadshacaWGObGaamyzaiaaykW7caaM % c8UaamyDaiaad6gacaWGPbGaamiDaiaaykW7caaMc8Uaam4yaiaadw % gacaWGSbGaamiBaaaacaGGLaaabaGaaGPaVlaaykW7caaMc8UaaGPa % VlaaykW7caaMc8UaaGPaVlaaykW7caaMc8UaaGPaVlaaykW7caaMc8 % UaaGPaVlaaykW7caaMc8UaaGPaVlaaykW7caaMc8UaaGPaVlaaykW7 % caaMc8UaaGPaVlaaykW7caaMc8UaaGPaVlaaykW7caaMc8UaaGPaVl % aaykW7caaMc8UaaGPaVlaaykW7caaMc8UaaGPaVlaaykW7caaMc8Ua % aGPaVlaaykW7caaMc8UaaGPaVlaaykW7caaMc8Uaeyypa0ZaaSaaae % aadaWcaaqaaiaaisdaaeaacaaIZaaaaiabec8aWjaadkhadaahaaWc % beqaaiaaiodaaaaakeaacaaI4aGaamOCamaaCaaaleqabaGaaG4maa % aaaaGccqGHxdaTcaaIXaGaaGimaiaaicdacqGH9aqpdaWcaaqaaiab % ec8aWbqaaiaaiAdaaaGaey41aqRaaGymaiaaicdacaaIWaGaaiyjaa % qaaiaaykW7caaMc8UaaGPaVlaaykW7caaMc8UaaGPaVlaaykW7caaM % c8UaaGPaVlaaykW7caaMc8UaaGPaVlaaykW7caaMc8UaaGPaVlaayk % W7caaMc8UaaGPaVlaaykW7caaMc8UaaGPaVlaaykW7caaMc8UaaGPa % VlaaykW7caaMc8UaaGPaVlaaykW7caaMc8UaaGPaVlaaykW7caaMc8 % UaaGPaVlaaykW7caaMc8UaaGPaVlaaykW7caaMc8UaaGPaVlaaykW7 % caaMc8Uaeyypa0JaaGynaiaaikdacaGGUaGaaG4maiaaiAdacaGGLa % Gaeyypa0JaaGynaiaaikdacaGGUaGaaGinaiaacwcaaaaa!5272! \begin{array}{l} \therefore Packing\,\,efficiency = \frac{{Volume\,\,occupied\,\,by\,\,four\,\,spheres\,\,in\,the\,\,unit\,\,cell \times 100}}{{Total\,\,volume\,\,of\,\,the\,\,unit\,\,cell}}\% \\ \,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\, = \frac{{\frac{4}{3}\pi {r^3}}}{{8{r^3}}} \times 100 = \frac{\pi }{6} \times 100\% \\ \,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\,\, = 52.36\% = 52.4\% \end{array}\)
Thus, we may conclude that ccp and hcp structures have maximum packing efficiency.
Calculations I Unit Cell DImensions
From the unit cell dimensions, it is possible to calculate the volume of the unit cell. Knowing the density of the metal, we can calculate the mass of the atoms in the unit cell. The determination of the mass of a single atom gives an accurate method of determination of Avogadro constant. Suppose, edge length of a unit cell of a cubic crystal determined by X-ray diffraction is a, d the density of the solid substance and M the molar mass. In case of cubic crystal:
Volume of a unit cell = a3
Mass of the unit cell
= number of atoms in unit cell × mass of each atom = z × m
(Here z is the number of atoms present in one unit cell and m is the mass of a single atom)
Mass of an atom present in the unit cell:
\(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGaamyBaiabg2 % da9maalaaabaGaamytaaqaaiaad6eadaWgaaWcbaGaamyqaaqabaaa % aaaa!3A95! m = \frac{M}{{{N_A}}}\) (M is molar mass)
Therefore, density of the unit cell
\(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGceaqabeaadaWcaa % qaaiaad2gacaWGHbGaam4CaiaadohacaaMc8UaaGPaVlaaykW7caWG % VbGaamOzaiaaykW7caaMc8UaaGPaVlaadwhacaWGUbGaamyAaiaads % hacaaMc8UaaGPaVlaadogacaWGLbGaamiBaiaadYgaaeaacaWG2bGa % am4BaiaadYgacaWG1bGaamyBaiaadwgacaaMc8UaaGPaVlaad+gaca % WGMbGaaGPaVlaaykW7caWG1bGaamOBaiaadMgacaWG0bGaaGPaVlaa % ykW7caWGJbGaamyzaiaadYgacaWGSbaaaaqaaiabg2da9maalaaaba % GaamOEaiaac6cacaWGTbaabaGaamyyamaaCaaaleqabaGaaG4maaaa % aaGccqGH9aqpdaWcaaqaaiaadQhacaGGUaGaamytaaqaaiaadggada % ahaaWcbeqaaiaaiodaaaGccaGGUaGaamOtamaaBaaaleaacaWGbbaa % beaaaaGccaaMc8UaaGPaVlaad+gacaWGYbGaaGPaVlaaykW7caWGKb % Gaeyypa0ZaaSaaaeaacaWG6bGaamytaaqaaiaadggadaahaaWcbeqa % aiaaiodaaaGccaWGobWaaSbaaSqaaiaadgeaaeqaaaaaaaaa!8500! \begin{array}{l} \frac{{mass\,\,\,of\,\,\,unit\,\,cell}}{{volume\,\,of\,\,unit\,\,cell}}\\ = \frac{{z.m}}{{{a^3}}} = \frac{{z.M}}{{{a^3}.{N_A}}}\,\,or\,\,d = \frac{{zM}}{{{a^3}{N_A}}} \end{array}\)
Remember, the density of the unit cell is the same as the density of the substance. The density of the solid can always be determined by other methods. Out of the five parameters (d, z, M, a and NA), if any four are known, we can determine the fifth.
EXAMPLE 3
An element has a body-centred cubic (bcc) structure with a cell edge of 288 pm. The density of the element is 7.2 g/cm3. How many atoms are present in 208 g of the element?
SOLUTION
Volume of the unit cell = (288 pm)3
= (288×10-12 m)3 = (288×10-10 cm)3
= 2.39×10-23 cm3
Volume of 208 g of the element\(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaWaaSaaaeaaca % WGTbGaamyyaiaadohacaWGZbaabaGaamizaiaadwgacaWGUbGaam4C % aiaadMgacaWG0bGaamyEaaaacqGH9aqpdaWcaaqaaiaaikdacaaIWa % GaaGioaiaadEgaaeaacaaI3aGaaiOlaiaaikdacaWGNbGaam4yaiaa % d2gadaahaaWcbeqaaiabgkHiTiaaiodaaaaaaOGaeyypa0JaaGOmai % aaiIdacaGGUaGaaGioaiaaiIdacaaMc8Uaam4yaiaad2gadaahaaWc % beqaaiaaiodaaaaaaa!548A! \frac{{mass}}{{density}} = \frac{{208g}}{{7.2gc{m^{ - 3}}}} = 28.88\,c{m^3}\)
Number of unit cells in this volume
\(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaWaaSaaaeaaca % aIYaGaaGioaiaac6cacaaI4aGaaGioaiaadogacaWGTbWaaWbaaSqa % beaacaaIZaaaaaGcbaGaaGOmaiaac6cacaaIZaGaaGyoaiabgEna0k % aaigdacaaIWaWaaWbaaSqabeaacqGHsislcaaIYaGaaG4maaaakiaa % ykW7caWGJbGaamyBamaaCaaaleqabaGaaG4maaaakiaac+cacaWG1b % GaamOBaiaadMgacaWG0bGaaGPaVlaaykW7caWGJbGaamyzaiaadYga % caWGSbaaaiabg2da9iaaigdacaaIYaGaaiOlaiaaicdacaaI4aGaey % 41aqRaaGymaiaaicdadaahaaWcbeqaaiaaikdacaaIZaaaaOGaaGPa % VlaaykW7caaMc8UaamyDaiaad6gacaWGPbGaamiDaiaaykW7caaMc8 % Uaam4yaiaadwgacaWGSbGaamiBaiaadohaaaa!6F67! =\frac{{28.88c{m^3}}}{{2.39 \times {{10}^{ - 23}}\,c{m^3}/unit\,\,cell}} = 12.08 \times {10^{23}}\,\,\,unit\,\,cells\)
Since each bcc cubic unit cell contains 2 atoms, therefore, the total number of atoms in 208 g = 2 (atoms/unit cell) × 12.08 × 1023 unit cells
= 24.16×1023 atoms
EXAMPLE 4
X-ray diffraction studies show that copper crystallises in an fcc unit cell with cell edge of 3.608×10-8 cm. In a separate experiment, copper is determined to have a density of 8.92 g/cm3, calculate the atomic mass of copper.
SOLUTION
In case of fcc lattice, number of atoms per unit cell, z = 4 atoms
Therefore, M = \(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaWaaSaaaeaaca % WGKbGaamOtamaaBaaaleaacaWGbbaabeaakiaadggadaahaaWcbeqa % aiaaiodaaaaakeaacaWG6baaaaaa!3B97! \frac{{d{N_A}{a^3}}}{z}\)
\(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGceaqabeaacqGH9a % qpdaWcaaqaaiaaiIdacaGGUaGaaGyoaiaaikdacaWGNbGaam4yaiaa % d2gadaahaaWcbeqaaiabgkHiTiaaiodaaaGccqGHxdaTcaaI2aGaai % OlaiaaicdacaaIYaGaaGOmaiabgEna0kaaigdacaaIWaWaaWbaaSqa % beaacaaIYaGaaG4maaaakiaadggacaWG0bGaam4Baiaad2gacaWGZb % GaaGPaVlaaykW7caWGTbGaam4BaiaadYgadaahaaWcbeqaaiabgkHi % TiaaigdaaaGccqGHxdaTdaqadaqaaiaaiodacaGGUaGaaGOnaiaaic % dacaaI4aGaey41aqRaaGymaiaaicdadaahaaWcbeqaaiabgkHiTiaa % iIdaaaGccaWGJbGaamyBaaGaayjkaiaawMcaamaaCaaaleqabaGaaG % 4maaaaaOqaaiaaisdacaaMc8UaaGPaVlaadggacaWG0bGaam4Baiaa % d2gacaWGZbaaaaqaaiabg2da9iaaiAdacaaIZaGaaiOlaiaaigdaca % aMc8UaaGPaVlaadEgacaGGVaGaamyBaiaad+gacaWGSbaaaaa!79C5! \begin{array}{l} = \frac{{8.92gc{m^{ - 3}} \times 6.022 \times {{10}^{23}}atoms\,\,mo{l^{ - 1}} \times {{\left( {3.608 \times {{10}^{ - 8}}cm} \right)}^3}}}{{4\,\,atoms}}\\ = 63.1\,\,g/mol \end{array}\)
Atomic mass of copper = 63.1u
EXAMPLE 5
Silver forms ccp lattice and X-ray studies of its crystals show that the edge length of its unit cell is 408.6 pm. Calculate the density of silver (Atomic mass = 107.9 u).
SOLUTION
Since the lattice is ccp, the number of silver atoms per unit cell = z = 4
Molar mass of silver = 107.9 g mol–1 = 107.9×10-3 kg mol–1
Edge length of unit cell = a = 408.6 pm = 408.6×10–12 m
Density,
\(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGceaqabeaacaWGKb % Gaeyypa0ZaaSaaaeaacaWG6bGaaiOlaiaad2eaaeaacaWGHbWaaWba % aSqabeaacaaIZaaaaOGaamOtamaaBaaaleaacaWGbbaabeaaaaaake % aacqGH9aqpdaWcaaqaaiaaisdacqGHxdaTdaqadaqaaiaaigdacaaI % WaGaaG4naiaac6cacaaI5aGaey41aqRaaGymaiaaicdadaahaaWcbe % qaaiabgkHiTiaaiodaaaGccaWGRbGaam4zaiaad2gacaWGVbGaamiB % amaaCaaaleqabaGaeyOeI0IaaGymaaaaaOGaayjkaiaawMcaaaqaam % aabmaabaGaaGinaiaaicdacaaI4aGaaiOlaiaaiAdacqGHxdaTcaaI % XaGaaGimamaaCaaaleqabaGaeyOeI0IaaGymaiaaikdaaaGccaWGTb % aacaGLOaGaayzkaaWaaWbaaSqabeaacaaIZaaaaOWaaeWaaeaacaaI % 2aGaaiOlaiaaicdacaaIYaGaaGOmaiabgEna0kaaigdacaaIWaWaaW % baaSqabeaacaaIYaGaaG4maaaakiaad2gacaWGVbGaamiBamaaCaaa % leqabaGaeyOeI0IaaGymaaaaaOGaayjkaiaawMcaaaaacqGH9aqpca % aIXaGaaGimaiaac6cacaaI1aGaey41aqRaaGymaiaaicdadaahaaWc % beqaaiaaiodaaaGccaWGRbGaam4zaiaad2gadaahaaWcbeqaaiabgk % HiTiaaiodaaaaakeaacqGH9aqpcaaIXaGaaGimaiaac6cacaaI1aGa % am4zaiaadogacaWGTbWaaWbaaSqabeaacqGHsislcaaIZaaaaaaaaa!854C! \begin{array}{l} d = \frac{{z.M}}{{{a^3}{N_A}}}\\ = \frac{{4 \times \left( {107.9 \times {{10}^{ - 3}}kgmo{l^{ - 1}}} \right)}}{{{{\left( {408.6 \times {{10}^{ - 12}}m} \right)}^3}\left( {6.022 \times {{10}^{23}}mo{l^{ - 1}}} \right)}} = 10.5 \times {10^3}kg{m^{ - 3}}\\ = 10.5gc{m^{ - 3}} \end{array}\)
INTEXT QUESTION
What is the two dimensional coordination number of a molecule in square close-packed layer?
A compound forms hexagonal close-packed structure. What is the total number of voids in 0.5 mol of it? How many of these are tetrahedral voids?
A compound is formed by two elements M and N. The element N forms ccp and atoms of M occupy 1/3rd of tetrahedral voids. What is the formula of the compound?
Which of the following lattices has the highest packing efficiency (i) simple cubic (ii) body-centred cubic and (iii) hexagonal close-packed lattice?
An element with molar mass 2.7×10-2 kg mol-1 forms a cubic unit cell with edge length 405 pm. If its density is 2.7×103 kg m-3, what is the nature of the cubic unit cell?
Imperfections in Solids
Although crystalline solids have short range as well as long range order in the arrangement of their constituent particles, yet crystals are not perfect. Usually a solid consists of an aggregate of large number of small crystals. These small crystals have defects in them. This happens when crystallisation process occurs at fast or moderate rate. Single crystals are formed when the process of crystallisation occurs at extremely slow rate. Even these crystals are not free of defects. The defects are basically irregularities in the arrangement of constituent particles. Broadly speaking, the defects are of two types, namely, point defects and line defects. Point defects are the irregularities or deviations from ideal arrangement around a point or an atom in a crystalline substance, whereas the line defects are the irregularities or deviations from ideal arrangement in entire rows of lattice points. These irregularities are called crystal defects. We shall confine our discussion to point defects only.
TYPES OF POINT DEFECT
Point defects can be classified into three types : (i) stoichiometric defects
(ii) impurity defects and (iii) non-stoichiometric defects.
(a) Stoichiometric Defects
These are the point defects that do not disturb the stoichiometry of the solid. They are also called intrinsic or thermodynamic defects. Basically these are of two types, vacancy defects and interstitial defects.
1. Vacancy Defect: When some of the lattice sites are vacant, the crystal is said to have vacancy defect (Fig. 1.27). This results in decrease in density of the substance. This defect can also develop when a substance is heated.

Fig. 1.27: Vacancy defects
(2) Interstitial Defect: When some constituent particles (atoms or molecules) occupy an interstitial site, the crystal is said to have interstitial defect (Fig. 1.28). This defect increases the density of the substance.
Fig. 1.28: Interstitial defects
Vacancy and interstitial defects as explained above can be shown by non-ionic solids. Ionic solids must always maintain electrical neutrality. Rather than simple vacancy or interstitial defects, they show these defects as Frenkel and Schottky defects.
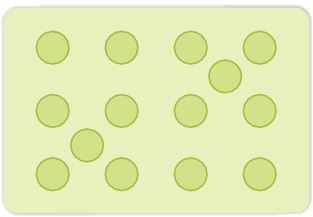
Fig. 1.28: Interstitial defects
(3) Frenkel Defect: This defect is shown by ionic solids. The smaller ion (usually cation) is dislocated from its normal site to an interstitial site (Fig. 1.29).

Fig. 1.29: Frenkel defects
It creates a vacancy defect at its original site and an interstitial defect at its new location.
Frenkel defect is also called dislocation defect.
It does not change the density of the solid. Frenkel defect is shown by ionic substance in which there is a large difference in the size of ions, for example, ZnS, AgCl, AgBr and AgI due to small size of Zn2+ and Ag+ ions.
(4) Schottky Defect: It is basically a vacancy defect in ionic solids. In order to maintain electrical neutrality, the number of missing cations and anions are equal (Fig. 1.30).

Fig. 1.30: Schottky defects
Like simple vacancy defect, Schottky defect also decreases the density of the substance. Number of such defects in ionic solids is quite significant. For example, in NaCl there are approximately 106 Schottky pairs per cm3 at room temperature. In 1 cm3 there are about 1022 ions. Thus, there is one Schottky defect per 1016 ions. Schottky defect is shown by ionic substances in which the cation and anion are of almost similar sizes. For example, NaCl, KCl, CsCl and AgBr. It may be noted that AgBr shows both, Frenkel as well as Schottky defects.
(b) Impurity Defects
If molten NaCl containing a little amount of SrCl2 is crystallised, some of the sites of Na+ ions are occupied by Sr2+ (Fig.1.31). Each Sr2+ replaces two Na+ ions. It occupies the site of one ion and the other site remains vacant. The cationic vacancies thus produced are equal in number to that of Sr2+ ions. Another similar example is the solid solution of CdCl2 and AgCl.
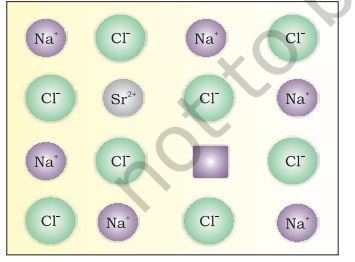
Fig. 1.31: Introduction of cation vacancy in NaCl by substitution of Na+ by Sr2+
(c) Non-Stoichiometric Defects
The defects discussed so far do not disturb the stoichiometry of the crystalline substance. However, a large number of non- stoichiometric inorganic solids are known which contain the constituent elements in non-stoichiometric ratio due to defects in their crystal structures. These defects are of two types: (i) metal excess defect and (ii) metal deficiency defect.
(1) Metal Excess Defect
Metal excess defect due to anionic vacancies: Alkali halides like NaCl and KCl show this type of defect. When crystals of NaCl are heated in an atmosphere of sodium vapour, the sodium atoms are deposited on the surface of the crystal.
The Cl– ions diffuse to the surface of the crystal and combine with Na atoms to give NaCl. This happens by loss of electron by sodium atoms to form Na+ ions. The released electrons diffuse into the crystal and occupy anionic sites (Fig. 1.32).
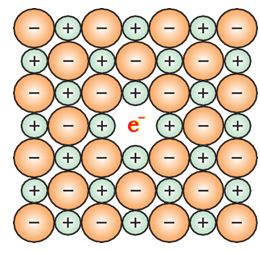
Fig. 1.32: An F-centre in a crystal
As a result the crystal now has an excess of sodium. The anionic sites occupied by unpaired electrons are called F-centres (from the German word Farbenzenter for colour centre). They impart yellow colour to the crystals of NaCl. The colour results by excitation of these electrons when they absorb energy from the visible light falling on the crystals. Similarly, excess of lithium makes LiCl crystals pink and excess of potassium makes KCl crystals violet (or lilac).
Metal excess defect due to the presence of extra cations at interstitial sites: Zinc oxide is white in colour at room temperature. On heating it loses oxygen and turns yellow.
ZnO
 \(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGaamOwaiaad6 % gadaahaaWcbeqaaiaaikdaaaGccqGHRaWkdaWcaaqaaiaaigdaaeaa % caaIYaaaaiaad+eadaWgaaWcbaGaaGOmaaqabaGccqGHRaWkcaaIYa % GaamyzamaaCaaaleqabaGaeyOeI0caaaaa!408C! Z{n^2} + \frac{1}{2}{O_2} + 2{e^ - }\)
\(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGaamOwaiaad6 % gadaahaaWcbeqaaiaaikdaaaGccqGHRaWkdaWcaaqaaiaaigdaaeaa % caaIYaaaaiaad+eadaWgaaWcbaGaaGOmaaqabaGccqGHRaWkcaaIYa % GaamyzamaaCaaaleqabaGaeyOeI0caaaaa!408C! Z{n^2} + \frac{1}{2}{O_2} + 2{e^ - }\)Now there is excess of zinc in the crystal and its formula becomes Zn O. The excess Zn2+ ions move to interstitial sites and the electrons to neighbouring interstitial sites.
(2) Metal Deficiency Defect
There are many solids which are difficult to prepare in the stoichiometric composition and contain less amount of the metal as compared to the stoichiometric proportion. A typical example of this type is FeO which is mostly found with a composition of Fe0.95O. It may actually range from Fe0.93O to Fe0.96O. In crystals of FeO some Fe2+ cations are missing and the loss of positive charge is made up by the presence of required number of Fe3+ ions.
ElectricalProperties
Solids exhibit an amazing range of electrical conductivities, extending over 27 orders of magnitude ranging from 10–20 to 107 ohm–1 m–1. Solids can be classified into three types on the basis of their conductivities.
(1) Conductors: The solids with conductivities ranging between 104 to 107 ohm–1m–1 are called conductors. Metals have conductivities in the order of 107 ohm–1m–1 are good conductors.
(2) Insulators : These are the solids with very low conductivities ranging between 10–20 to 10–10 ohm–1m–1.
(3) Semiconductors : These are the solids with conductivities in the intermediate range from 10–6 to 104 ohm–1m–1.
Conduction OF Electricity in MetalS
A conductor may conduct electricity through movement of electrons or ions. Metallic conductors belong to the former category and electrolytes to the latter.
Metals conduct electricity in solid as well as molten state. The conductivity of metals depend upon the number of valence electrons available per atom. The atomic orbitals of metal atoms form molecular orbitals which are so close in energy to each other as to form a band. If this band is partially filled or it overlaps with a higher energy unoccupied conduction band, then electrons can flow easily under an applied electric field and the metal shows conductivity (Fig. 1.33 a).
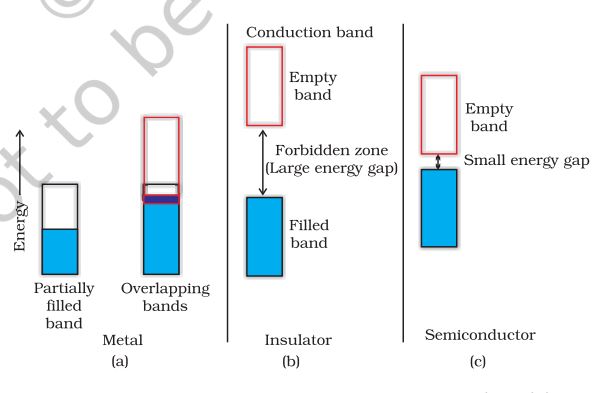
Fig. 1.33
Distinction among
(a) metals
(b) insulators and
(c) semiconductors. In each case, an unshaded area represents a conduction band.
If the gap between filled valence band and the next higher unoccupied band (conduction band) is large, electrons cannot jump to it and such a substance has very small conductivity and it behaves as an insulator (Fig. 1.33 b).
Conduction OF Electricity in Semi- conductors
In case of semiconductors, the gap between the valence band and conduction band is small (Fig. 1.33 c). Therefore, some electrons may jump to conduction band and show some conductivity. Electrical conductivity of semiconductors increases with rise in temperature, since more electrons can jump to the conduction band. Substances like silicon and germanium show this type of behaviour and are called intrinsic semiconductors.
The conductivity of these intrinsic semiconductors is too low to be of practical use. Their conductivity is increased by adding an appropriate amount of suitable impurity. This process is called doping. Doping can be done with an impurity which is electron rich or electron deficient as compared to the intrinsic semiconductor silicon or germanium. Such impurities introduce electronic defects in them.
(a) Electron – rich impurities
Silicon and germanium belong to group 14 of the periodic table and have four valence electrons each. In their crystals each atom forms four covalent bonds with its neighbours (Fig. 1.34 a). When doped with a group 15 element like P or As, which contains five valence electrons, they occupy some of the lattice sites in silicon or germanium crystal (Fig. 1.34 b).
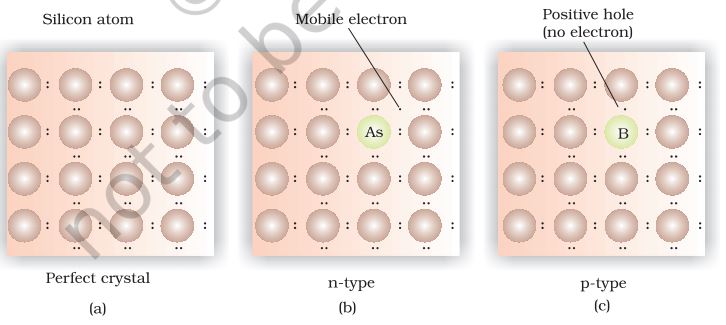
Fig. 1.34: Creation of n-type and p-type semiconductors by doping groups 13 and 15 elements.
Four out of five electrons are used in the formation of four covalent bonds with the four neighbouring silicon atoms. The fifth electron is extra and becomes delocalised. These delocalised electrons increase the conductivity of doped silicon (or germanium). Here the increase in conductivity is due to the negatively charged electron, hence silicon doped with electron-rich impurity is called n-type semiconductor.
(b) Electron – deficit impurities
Silicon or germanium can also be doped with a group 13 element like B, Al or Ga which contains only three valence electrons. The place where the fourth valence electron is missing is called electron hole or electron vacancy (Fig. 1.34 c). An electron from a neighbouring atom can come and fill the electron hole, but in doing so it would leave an electron hole at its original position. If it happens, it would appear as if the electron hole has moved in the direction opposite to that of the electron that filled it. Under the influence of electric field, electrons would move towards the positively charged plate through electronic holes, but it would appear as if electron holes are positively charged and are moving towards negatively charged plate. This type of semi conductors are called p-type semiconductors.
Applications of n-type and p-type semiconductors
Various combinations of n-type and p-type semiconductors are used for making electronic components. Diode is a combination of n-type and p-type semiconductors and is used as a rectifier. Transistors are made by sandwiching a layer of one type of semiconductor between two layers of the other type of semiconductor. npn and pnp type of transistors are used to detect or amplify radio or audio signals. The solar cell is an efficient photo-diode used for conversion of light energy into electrical energy.
Germanium and silicon are group 14 elements and therefore, have a characteristic valence of four and form four bonds as in diamond. A large variety of solid state materials have been prepared by combination of groups 13 and 15 or 12 and 16 to simulate average valence of four as in Ge or Si. Typical compounds of groups 13 – 15 are InSb, AlP and GaAs. Gallium arsenide (GaAs) semiconductors have very fast response and have revolutionised the design of semiconductor devices. ZnS, CdS, CdSe and HgTe are examples of groups 12 – 16 compounds. In these compounds, the bonds are not perfectly covalent and the ionic character depends on the electronegativities of the two elements.
It is interesting to learn that transition metal oxides show marked differences in electrical properties. TiO, CrO2 and ReO3 behave like metals. Rhenium oxide, ReO3 is like metallic copper in its conductivity and appearance. Certain other oxides like VO, VO2, VO3 and TiO3 show metallic or insulating properties depending on temperature.
Magnetic Properties
Every substance has some magnetic properties associated with it. The origin of these properties lies in the electrons. Each electron in an atom behaves like a tiny magnet. Its magnetic moment originates from two types of motions (i) its orbital motion around the nucleus and (ii) its spin around its own axis (Fig. 1.35).
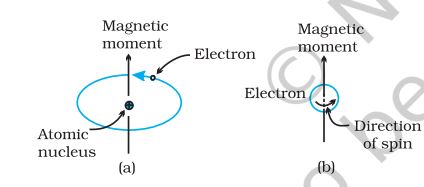
Fig.1.35: Demonstration of the magnetic moment associated with (a) an orbiting electron and (b) a spinning electron.
Electron being a charged particle and undergoing these motions can be considered as a small loop of current which possesses a magnetic moment. Thus, each electron has a permanent spin and an orbital magnetic moment associated with it. Magnitude of this magnetic moment is very small and is measured in the unit called Bohr magneton, \(\mu_B\) . It is equal to 9.27 × 10–24A m2. On the basis of their magnetic properties, substances can be classified into five categories: (i) paramagnetic (ii) diamagnetic
(iii)ferromagnetic (iv) antiferromagnetic and (v) ferrimagnetic.
(1) Paramagnetism: Paramagnetic substances are weakly attracted by a magnetic field. They are magnetised in a magnetic field in the same direction. They lose theirmagnetism in the absence of magnetic field. Paramagnetism is due to presence of one or more unpaired electrons which are attracted by the magnetic field. O , Cu2+, Fe3+, Cr3+ are some examples of such substances.
(2) Diamagnetism: Diamagnetic substances are weakly repelled by a magnetic field. H2O, NaCl and C6H6 are some examples of such substances. They are weakly magnetised in a magnetic field in opposite direction. Diamagnetism is shown by those substances in which all the electrons are paired and there are no unpaired electrons. Pairing of electrons cancels their magnetic moments and they lose their magnetic character.
(3) Ferromagnetism: A few substances like iron, cobalt, nickel, gadolinium and CrO2 are attracted very strongly by a magnetic field. Such substances are called ferromagnetic substances. Besides strong attractions, these substances can be permanently magnetised. In solid state, the metal ions of ferromagnetic substances are grouped together into small regions called domains. Thus, each domain acts as a tiny magnet. In an unmagnetised piece of a ferromagnetic substance the domains are randomly oriented and their magnetic moments get cancelled. When the substance is placed in a magnetic field all the domains get oriented in the direction of the magnetic field (Fig. 1.36 a) and a strong magnetic effect is produced. This ordering of domains persist even when the magnetic field is removed and the ferromagnetic substance becomes a permanent magnet.

Fig 1.36: Schematic alignment of magnetic moments in (a) ferromagnetic (b) antiferromagnetic and (c) ferrimagnetic.
(4) Antiferromagnetism: Substances like MnO showing anti- ferromagnetism have domain structure similar to ferromagnetic substance, but their domains are oppositely oriented and cancel out each other's magnetic moment (Fig. 1.36 b).
(5) Ferrimagnetism: Ferrimagnetism is observed when the magnetic moments of the domains in the substance are aligned in parallel and anti-parallel directions in unequal numbers (Fig. 1.36c). They are weakly attracted by magnetic field as compared to ferromagnetic substances. Fe3O4 (magnetite) and ferrites like MgFe2O4 and ZnFe2O4 are examples of such substances. These substances also lose ferrimagnetism on heating and become paramagnetic.
INTEXT QUESTION
What type of defect can arise when a solid is heated? Which physical property is affected by it and in what way?
What type of stoichiometric defect is shown by:
(1) ZnS (2) AgBr
Explain how vacancies are introduced in an ionic solid when a cation of higher valence is added as an impurity in it.
Ionic solids, which have anionic vacancies due to metal excess defect, develop colour. Explain with the help of a suitable example.
A group 14 element is to be converted into n-type semiconductor by doping it with a suitable impurity. To which group should this impurity belong?
What type of substances would make better permanent magnets, ferromagnetic or ferrimagnetic. Justify your answer.
-
Haloalkanes and Haloarenes . 10
The replacement of hydrogen atom(s) in an aliphatic or aromatic hydrocarbon by halogen atom(s) results in the formation of alkyl halide (haloalkane) and aryl halide (haloarene), respectively. Haloalkanes contain halogen atom(s) attached to the sp3 hybridised carbon atom of an alkyl group whereas haloarenes contain halogen atom(s) attached to sp2 hybridised carbon atom(s) of an aryl group. Many halogen containing organic compounds occur in nature and some of these are clinically useful. These classes of compounds find wide applications in industry as well as in day- to-day life. They are used as solvents for relatively non-polar compounds and as starting materials for the synthesis of wide range of organic compounds. Chlorine containing antibiotic, chloramphenicol, produced by microorganisms is very effective for the treatment of typhoid fever. Our body produces iodine containing hormone, thyroxine, the deficiency of which causes a disease called goiter. Synthetic halogen compounds, viz. chloroquine is used for the treatment of malaria; halothane is used as an anaesthetic during surgery. Certain fully fluorinated compounds are being considered as potential blood substitutes in surgery.
In this Unit, you will study the important methods of preparation, physical and chemical properties and uses of organohalogen compounds.
Classification
Haloalkanes and haloarenes may be classified as follows:
On THE Basis of Number of Halogen Atoms
These may be classified as mono, di, or polyhalogen (tri-,tetra-, etc.) compounds depending on whether they contain one, two or more halogen atoms in their structures. For example,
Monohalocompounds may further be classified according to the hybridisation of the carbon atom to which the halogen is bonded, as discussed below.
Compounds Containing sp3 C—X Bond (X= F, Cl, Br, I)
(a) Alkyl halides or haloalkanes (R—X)
In alkyl halides, the halogen atom is bonded to an alkyl group (R). They form a homologous series represented by CnH2n+1X. They are further classified as primary, secondary or tertiary according to the nature of carbon to which halogen is attached. If halogen is attached to a primary carbon atom in an alkyl halide, the alkyl halide is called primary alkyl halide or 1° alkyl halide. Similarly, if halogen is attached to secondary or tertiary carbon atom, the alkyl halide is called secondary alkyl halide (2°) and tertiary (3°) alkyl halide, respectively.
(b) Allylic halides
These are the compounds in which the halogen atom is bonded to an sp3-hybridised carbon atom adjacent to carbon-carbon double bond (C=C) i.e. to an allylic carbon.

(c) Benzylic halides
These are the compounds in which the halogen atom is bonded to an sp3-hybridised carbon atom attached to an aromatic ring.
This class includes:
Compounds Containing sp2 C—X Bond
(a) Vinylic halides
These are the compounds in which the halogen atom is bonded to a sp2-hybridised carbon atom of a carbon-carbon double bond
(b) Aryl halides
These are the compounds in which the halogen atom is directly bonded to the sp2-hybridised carbon atom of an aromatic ring.
Nomenclature
Having learnt the classification of halogenated compounds, let us now learn how these are named. The common names of alkyl halides are derived by naming the alkyl group followed by the name of halide. In the IUPAC system of nomenclature, alkyl halides are named as halosubstituted hydrocarbons. For mono halogen substituted derivatives of benzene, common and IUPAC names are the same. For dihalogen derivatives, the prefixes o-, m-, p- are used in common system but in IUPAC system, as you have learnt in Class XI, Unit 12, the numerals 1,2; 1,3 and 1,4 are used.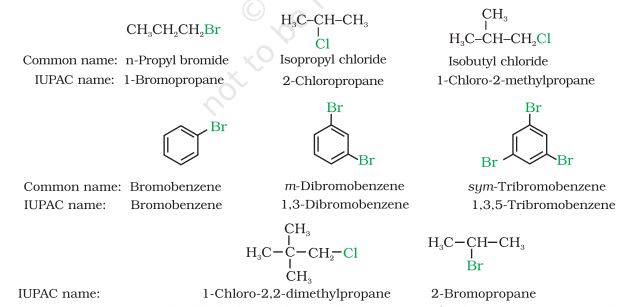
The dihaloalkanes having the same type of halogen atoms are named as alkylidene or alkylene dihalides. The dihalo-compounds having both the halogen atoms are further classified as geminal halides or gem-dihalides when both the halogen atoms are present on the same carbon atom of the chain and vicinal halides or vic-dihalides when halogen atoms are present on adjacent carbon atoms. In common name system, gem-dihalides are named as alkylidene halides and vic-dihalides are named as alkylene dihalides. In IUPAC system, they are named as dihaloalkanes.

Some common examples of halocompounds are mentioned in Table 10.1.
Table 10.1: Common and IUPAC Names of some Halides
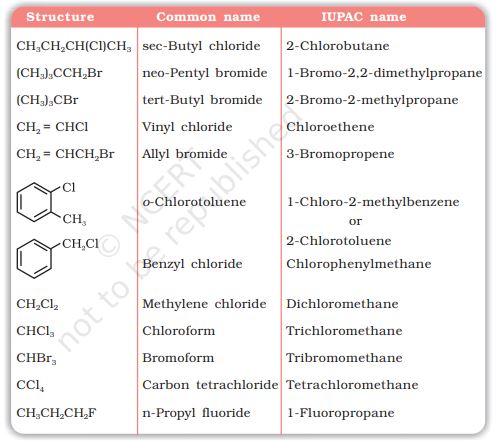
EXAMPLE 1
Draw the structures of all the eight structural isomers that have the molecular formula C5H11Br. Name each isomer according to IUPAC system and classify them as primary, secondary or tertiary bromide.
SOLUTION
CH3CH2CH2CH2Br 1-Bromopentane (1o)
CH3CH2CH2CH(Br)CH3 2-Bromopentane(2o)
CH3CH2CH(Br)CH2CH 3-Bromopentane (2o)
(CH3)2CHCH2CH2Br 1-Bromo-3-methylbutane (1o)
(CH3)2CHCHBrCH3 2-Bromo-3-methylbutane(2o)
(CH3)2CBrCH2CH3 2-Bromo-2-methylbutane (3o)
CH3CH2CH(CH3)CH2Br 1-Bromo-2-methylbutane(1o)
(CH3)3CCH2Br 1-Bromo-2,2-dimethylpropane (1o)
EXAMPLE 2
Write IUPAC names of the following:
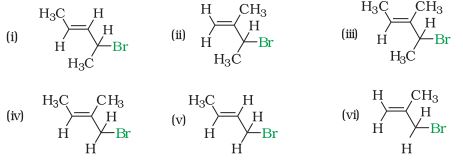
(i) 4-Bromopent-2-ene
(ii) 3-Bromo-2-methylbut-1-ene
(iii) 4-Bromo-3-methylpent-2-ene
(iv) 1-Bromo-2-methylbut-2-ene
(vi) 3-Bromo-2-methylpropene
(v) 1-Bromobut-2-ene
INTEXT QUESTION
Write structures of the following compounds:
1. 1-Bromo-4-sec. butyl-2-methylbenzene.
2. 1,4-Dibromobut-2-ene
3. 4-tert. Butyl-3-iodoheptane
4. 1-Chloro-4-ethylcyclohexane
5. 2-Chloro-3-methylpentane
Nature OF C-X Bond
Halogen atoms are more electronegative than carbon, therefore, carbon-halogen bond of alkyl halide is polarised; the carbon atom bears a partial positive charge whereas the halogen atom bears a partial negative charge.
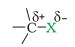
As we go down the group in the periodic table, the size of halogen atom increases. Fluorine atom is the smallest and iodine atom is the largest. Consequently the carbon-halogen bond length also increases from C—F to C—I. Some typical bond lengths, bond enthalpies and dipole moments are given in Table 10.2.
Alkyl halides are best prepared from alcohols, which are easily accessible.
Table 10.2: Carbon-Halogen (C—X) Bond Lengths, Bond Enthalpies and Dipole Moments
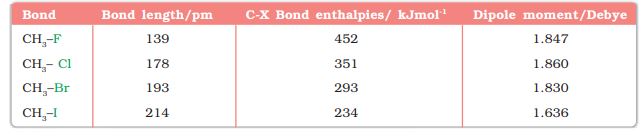
Methods OF PreparatioN of Haloalkanes
From Alcohols
The hydroxyl group of an alcohol is replaced by halogen on reaction with concentrated halogen acids, phosphorus halides or thionyl chloride. Thionyl chloride is preferred because in this reaction alkyl halide is formed along with gases SO2 and HCl. The two gaseous products are escapable,hence, the reaction gives pure alkyl halides. The reactions of primary and secondary alcohols with HCl require the presence of a catalyst, ZnCl2. With tertiary alcohols, the reaction is conducted by simply shaking the alcohol with concentrated HCl at room temperature. Constant boiling with HBr (48%) is used for preparing alkyl bromide. Good yields of R—I may be obtained by heating alcohols with sodium or potassium iodide in 95% orthophosphoric acid. The order of reactivity of alcohols with a given haloacid is 3°>2°>1°. Phosphorus tribromide and triiodide are usually generated in situ (produced in the reaction mixture) by the reaction of red phosphorus with bromine and iodine respectively.
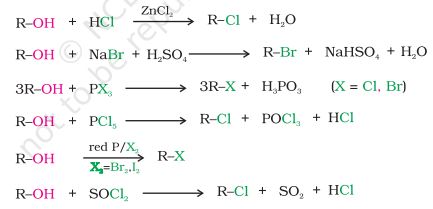
The preparation of alkyl chloride is carried out either by passing dry hydrogen chloride gas through a solution of alcohol or by heating a mixture of alcohol and concentrated aqueous halogen acid.
The above methods are not applicable for the preparation of aryl halides because the carbon-oxygen bond in phenols has a partial double bond character and is difficult to break being stronger than a single bond (Unit 11, Class XI).
From Hydrocarbons
(I) From alkanes by free radical halogenation
Free radical chlorination or bromination of alkanes gives a complex mixture of isomeric mono- and polyhaloalkanes, which is difficult to separate as pure compounds. Consequently, the yield of any single compound is low (Unit 13, Class XI).
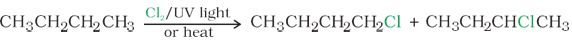
(II) From alkenes
a. Addition of hydrogen halides: An alkene is converted to corresponding alkyl halide by reaction with hydrogen chloride, hydrogen bromide or hydrogen iodide.
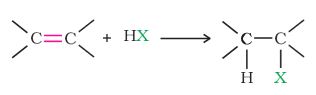
Propene yields two products, however only one predominates as per Markovnikov’s rule. (Unit 13, Class XI)
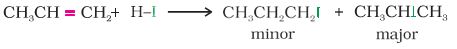
b. Addition of halogens: In the laboratory, addition of bromine in CCl4 to an alkene resulting in discharge of reddish brown colour of bromine constitutes an important method for the detection of double bond in a molecule. The addition results in the synthesis of vic-dibromides, which are colourless (Unit 13, Class XI).
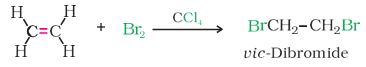
EXAMPLE 3
Identify all the possible monochloro structural isomers expected to be formed on free radical monochlorination of (CH3)2CHCH2CH3.
SOLUTION
In the given molecule, there are four different types of hydrogen atoms.
Replacement of these hydrogen atoms will give the following
(CH3)2CHCH2CH2Cl (CH3)2CHCH(Cl)CH3
(CH3)2C(Cl)CH2CH3 CH3CH(CH2Cl)CH2CH3
Halogen Exchange
Alkyl iodides are often prepared by the reaction of alkyl chlorides/ bromides with NaI in dry acetone. This reaction is known as Finkelstein reaction.

NaCl or NaBr thus formed is precipitated in dry acetone. It facilitates the forward reaction according to Le Chatelier’s Principle.
The synthesis of alkyl fluorides is best accomplished by heating an alkyl chloride/bromide in the presence of a metallic fluoride such as AgF, Hg2F2, CoF2 or SbF3. The reaction is termed as Swarts reaction.

Preparation OF Haloarenes
(I) From hydrocarbons by electrophilic substitution
Aryl chlorides and bromides can be easily prepared by electrophilic substitution of arenes with chlorine and bromine respectively in the
presence of Lewis acid catalysts like iron or iron(III) chloride.
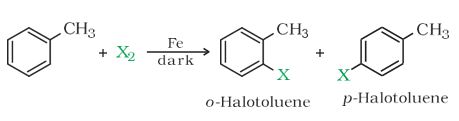
The ortho and para isomers can be easily separated due to large difference in their melting points. Reactions with iodine are reversible in nature and require the presence of an oxidising agent (HNO3, HIO4) to oxidise the HI formed during iodination. Fluoro compounds are not prepared by this method due to high reactivity of fluorine.
(II) From amines by Sandmeyer’s reaction
When a primary aromatic amine, dissolved or suspended in cold aqueous mineral acid, is treated with sodium nitrite, a diazonium salt is formed (Unit 13, Class XII). Mixing the solution of freshly prepared diazonium salt with cuprous chloride or cuprous bromide results in the replacement of the diazonium group by –Cl or –Br.

Replacement of the diazonium group by iodine does not require the presence of cuprous halide and is done simply by shaking the diazonium salt with potassium iodide.
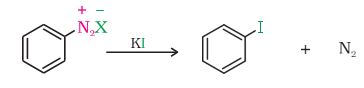
EXAMPLE 4
Write the products of the following reactions:
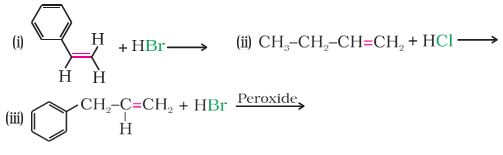
SOLUTION
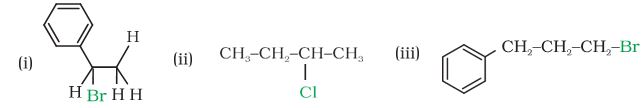
INTEXT QUESTION
Why is sulphuric acid not used during the reaction of alcohols with KI?
Why structures of different dihalogen derivatives of propane.
Among the isomeric alkanes of molecular formula C5H12, identify the one that on photochemical chlorination yields
A single monochloride.
Three isomeric monochlorides.
Four isomeric monochlorides.
Draw the structures of major monohalo products in each of the following reactions:

Physical Properties
Alkyl halides are colourless when pure. However, bromides and iodides develop colour when exposed to light. Many volatile halogen compounds have sweet smell.
Melting and boiling points
Methyl chloride, methyl bromide, ethyl chloride and some chlorofluoromethanes are gases at room temperature. Higher members are liquids or solids. As we have already learnt, molecules of organic halogen compounds are generally polar. Due to greater polarity as well as higher molecular mass as compared to the parent hydrocarbon, the intermolecular forces of attraction (dipole-dipole and van der Waals) are stronger in the halogen derivatives. That is why the boiling points of chlorides, bromides and iodides are considerably higher than those of the hydrocarbons of comparable molecular mass.
The attractions get stronger as the molecules get bigger in size and have more electrons. The pattern of variation of boiling points of different halides is depicted in Fig. 10.1.

Fig. 10.1: Comparison of boiling points of some alkyl halides
The boiling points of isomeric haloalkanes decrease with increase in branching (Unit 12, Class XI). For example, 2-bromo-2- methylpropane has the lowest boiling point among the three isomers.
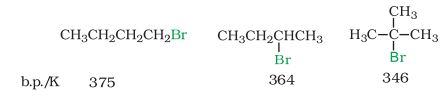
Boiling points of isomeric dihalobenzenes are very nearly the same. However, the para-isomers are high melting as compared to their ortho- and meta-isomers. It is due to symmetry of para-isomers that fits in crystal lattice better as compared to ortho- and meta-isomers.
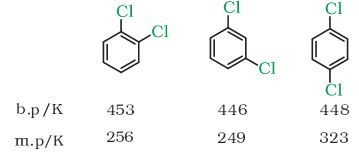
Density
Bromo, iodo and polychloro derivatives of hydrocarbons are heavier than water. The density increases with increase in number of carbon atoms, halogen atoms and atomic mass of the halogen atoms (Table 10.3).
Table 10.3: Density of Some Haloalkanes

Solubility
The haloalkanes are very slightly soluble in water. In order to dissolve haloalkane in water, energy is required to overcome the attractions between the haloalkane molecules and break the hydrogen bonds between water molecules. Less energy is released when new attractions are set up between the haloalkane and the water molecules as these are not as strong as the original hydrogen bonds in water. As a result, the solubility of haloalkanes in water is low. However, haloalkanes tend to dissolve in organic solvents because the new intermolecular attractions between haloalkanes and solvent molecules have much the same strength as the ones being broken in the separate haloalkane and solvent molecules.
INTEXT QUESTION
Arrange each set of compounds in order of increasing boiling points.
1. Bromomethane, Bromoform, Chloromethane, Dibromomethane.
2. 1-Chloropropane, Isopropyl chloride, 1-Chlorobutane.
Chemical ReactionS
The reactions of haloalkanes may be divided into the following categories:
1. Nucleophilic substitution
2. Reaction with metals.
3. Elimination reactions
Reactions OF Haloalkanes
1. Nucleophilic substitution reactions
You have learnt in Class XI that nucleophiles are electron rich species. Therefore, they attack at that part of the substrate molecule which is electron deficient. The reaction in which a nucleophile replaces already existing nucleophile in a molecule is called nucleophilic substitution reaction. Haloalkanes are substrate in these reactions. In this type of reaction, a nucleophile reacts with haloalkane (the substrate) having a partial positive charge on the carbon atom bonded to halogen. A substitution reaction takes place and halogen atom, called leaving group departs as halide ion. Since the substitution reaction is initiated by a nucleophile, it is called nucleophilic substitution reaction.

It is one of the most useful classes of organic reactions of alkyl halides in which halogen is bonded to sp3 hybridised carbon. The products formed by the reaction of haloalkanes with some common nucleophiles are given in Table 10.4.
Table 10.4: Nucleophilic Substitution of Alkyl Halides (R–X)
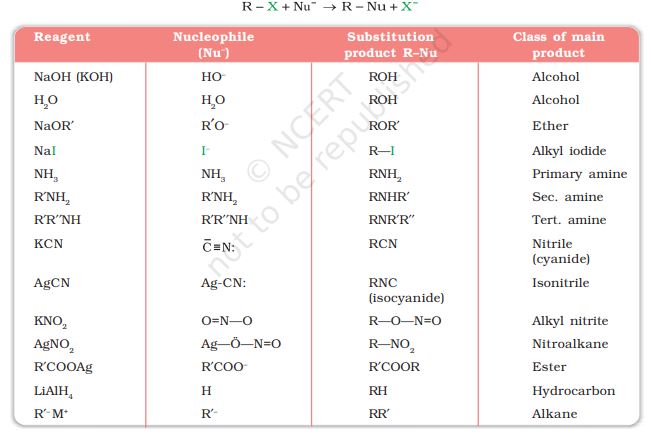
EXAMPLE 5
SOLUTION
KCN is predominantly ionic and provides cyanide ions in solution. Although both carbon and nitrogen atoms are in a position to donate electron pairs, the attack takes place mainly through carbon atom and not through nitrogen atom since C—C bond is more stable than C—N bond. However, AgCN is mainly covalent in nature and nitrogen is free to donate electron pair forming isocyanide as the main product.
Mechanism: This reaction has been found to proceed by two different mechanims which are described below:
(a) Substitution nucleophilic bimolecular (SN2)
The reaction between CH3Cl and hydroxide ion to yield methanol and chloride ion follows a second order kinetics, i.e., the rate depends upon the concentration of both the reactants.
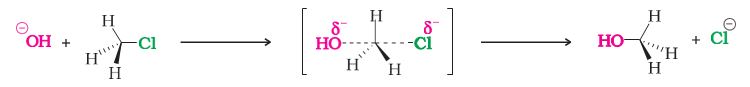
You have already learnt in Section 12.3.2 of Class XI, the solid wedge represents the bond coming out of the paper, dashed line going down the paper and a straight line representing bond in the plane of the paper.
The above reaction can be represented diagrammatically as shown in Fig. 10.2.
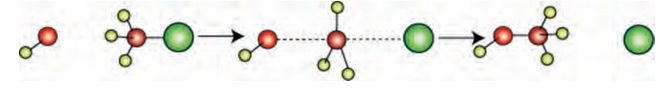
Fig. 10.2: Red ball represents the incoming hydroxide ion and green ball represents the outgoing halide ion
It depicts a bimolecular nucleophilic substitution (SN2) reaction; the incoming nucleophile interacts with alkyl halide causing the carbon-halide bond to break and a new bond is formed between carbon and attacking nucleophile. Here it is C-O bond formed between C and -OH. These two processes take place simultaneously in a single step and no intermediate is formed. As the reaction progresses and the bond between the incoming nucleophile and the carbon atom starts forming, the bond between carbon atom and leaving group weakens. As this happens, the three carbon-hydrogen bonds of the substrate start moving away from the attacking nucleophile. In transition state all the three C-H bonds are in the same plane and the attacking and leaving nucleophiles are partially attached to the carbon. As the attacking nucleophile approaches closer to the carbon, C-H bonds still keep on moving in the same direction till the attacking nucleophile attaches to carbon and leaving group leaves the carbon. As a result configuration is inverted, the configuration (See box) of carbon atom under attack inverts in much the same way as an umbrella is turned inside out when caught in a strong wind. This process is called as inversion of configuration. In the transition state, the carbon atom is simultaneously bonded to incoming nucleophile and the outgoing leaving group. Such structures are unstable and cannot be isolated. Thus, in the transition state, carbon is simultaneously bonded to five atoms.
Configuration
Spacial arrangement of functional groups around carbon is called its configuration. See the structures (A) and (B) given below carefully.
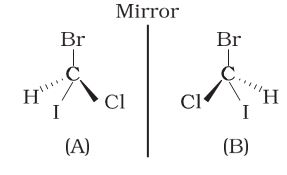
These are the two structures of the same compound. They differ in spacial arrangement of functional groups attached to carbon. Structure (A) is mirror image of Structure (B). We say configuration of carbon in structure (A) is mirror image of the configuration of carbon in structure (B).
Hughes worked under Ingold and earned a D.Sc. degree from the University of London.
Since this reaction requires the approach of the nucleophile to the carbon bearing the leaving group, the presence of bulky substituents on or near the carbon atom have a dramatic inhibiting effect. Of the simple alkyl halides, methyl halides react most rapidly in SN2 reactions because there are only three small hydrogen atoms. Tertiary halides are the least reactive because bulky groups hinder the approaching nucleophiles. Thus the order of reactivity followed is:
Primary halide > Secondary halide > Tertiary halide.

Fig.10.3: Steric effects in SN2 reaction. The relative rate of SN2 reaction is given in parenthesis
(b) Substitution nucleophilic unimolecular (SN1)
SN1 reactions are generally carried out in polar protic solvents (like water, alcohol, acetic acid, etc.). The reaction between tert- butyl bromide and hydroxide ion yields tert-butyl alcohol and follows the first order kinetics, i.e., the rate of reaction depends upon the concentration of only one reactant, which is tert- butyl bromide.
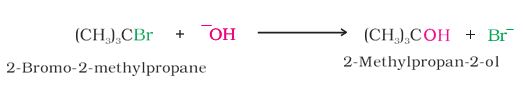
It occurs in two steps. In step I, the polarised C—Br bond undergoes slow cleavage to produce a carbocation and a bromide ion. The carbocation thus formed is then attacked by nucleophile in step II to complete the substitution reaction.
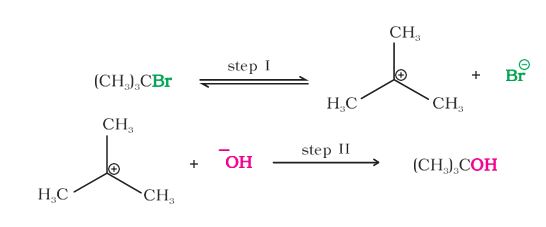
Step I is the slowest and reversible. It involves the C–Br bond breaking for which the energy is obtained through solvation of halide ion with the proton of protic solvent. Since the rate of reaction depends upon the slowest step, the rate of reaction depends only on the concentration of alkyl halide and not on the concentration of hydroxide ion. Further, greater the stability of carbocation, greater will be its ease of formation from alkyl halide and faster will be the rate of reaction. In case of alkyl halides, 30 alkyl halides undergo S 1 reaction very fast because of the high stability of 30 carbocations. We can sum up the order of reactivity of alkyl halides towards SN1 and SN2 reactions as follows:

For the same reasons, allylic and benzylic halides show high reactivity towards the SN1 reaction. The carbocation thus formed gets stabilised through resonance (Unit 12, Class XI) as shown below:
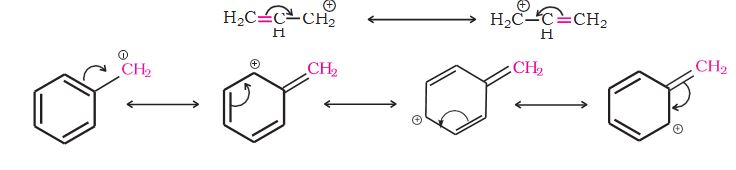
For a given alkyl group, the reactivity of the halide, R-X, follows the same order in both the mechanisms R–I> R–Br>R–Cl>>R–F.
EXAMPLE 6
In the following pairs of halogen compounds, which would undergo SN2 reaction faster?

SOLUTION
 It is primary halide and therefore undergoes SN2 reaction faster.
It is primary halide and therefore undergoes SN2 reaction faster. As iodine is a better leaving group because of its large size, it will be released at a faster rate in the presence of incoming nucleophile.
As iodine is a better leaving group because of its large size, it will be released at a faster rate in the presence of incoming nucleophile.EXAMPLE 7
Predict the order of reactivity of the following compounds in SN1 and SN2 reactions:
1. The four isomeric bromobutanes
2. C6H5CH2Br, C6H5CH(C6H5)Br, C6H5CH(CH3)Br, C6H5C(CH3)(C6H5)Br
SOLUTION
1. CH3CH2CH2CH2Br < (CH3)2CHCH2Br < CH3CH2CH(Br)CH3 < (CH3)3CBr (SN1) CH3CH2CH2CH2Br > (CH3)2CHCH2Br > CH3CH2CH(Br)CH3 > (CH3)3CBr (SN2) Of the two primary bromides, the carbocation intermediate derived from (CH3)2CHCH2Br is more stable than derived from CH3CH2CH2CH2Br because of greater electron donating inductive effect of (CH3)2CH- group. Therefore, (CH3)2CHCH2Br is more reactive than CH3CH2CH2CH2Br in SN1 reactions. CH3CH2CH(Br)CH3 is a secondary bromide and (CH3)3CBr is a tertiary bromide. Hence the above order is followed in SN1. The reactivity in SN2 reactions follows the reverse order as the steric hinderance around the electrophilic carbon increases in that order.
2. C6H5C(CH3)(C6H5)Br > C6H5CH(C6H5)Br > C6H5CH(CH3)Br > C6H5CH2Br (SN1)
C6H5C(CH3)(C6H5)Br < C6H5CH(C6H5)Br < C6H5CH(CH3)Br < C6H5CH2Br (SN2)
Of the two secondary bromides, the carbocation intermediate obtained from C6H5CH(C6H5)Br is more stable than obtained from C6H5CH(CH3)Br because it is stabilised by two phenyl groups due to resonance. Therefore, the former bromide is more reactive than the latter in SN1 reactions. A phenyl group is bulkier than a methyl group. Therefore, C6H5CH(C6H5)Br is less reactive than C
6H5CH(CH3)Br in SN2 reactions.c. Stereochemical aspects of nucleophilic substitution reactions In order to understand the stereochemical aspects of substitution reactions, we need to learn some basic stereochemical principles and notations (optical activity, chirality, retention, inversion, racemisation, etc.).
1. Optical activity: Plane of plane polarised light produced by passing ordinary light through Nicol prism is rotated when it is passed through the solutions of certain compounds. Such compounds are called optically active compounds. The angle by which the plane polarised light is rotated is measured by an instrument called polarimeter. If the compound rotates the plane of plane polarised light to the right, i.e., clockwise direction, it is called dextrorotatory (Greek for right rotating) or the d-form and is indicated by placing a positive (+) sign before the degree of rotation. If the light is rotated towards left (anticlockwise direction), the compound is said to be laevo-rotatory or the l-form and a negative (–) sign is placed before the degree of rotation. Such (+) and (–) isomers of a compound are called optical isomers and the phenomenon is termed as optical isomerism.
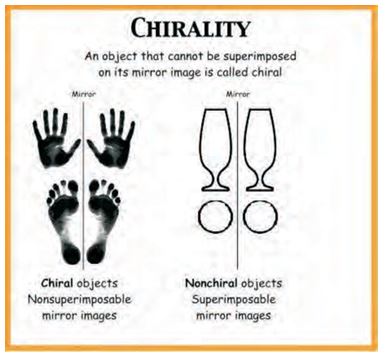
Fig 10.4: Some common examples of chiral andachiral objects
William Nicol (1768- 1851) developed the first prism that produced plane polarised light.
2. Molecular asymmetry, chirality and enantiomers: The observation of Louis Pasteur (1848) that crystals of certain compounds exist in the form of mirror images laid the foundation of modern stereochemistry. He demonstrated that aqueous solutions of both types of crystals showed optical rotation, equal in magnitude (for solution of equal concentration) but opposite in direction. He believed that this difference in optical activity was associated with the three dimensional arrangements of atoms in the molecules (configurations) of two types of crystals. Dutch scientist, J. Van’t Hoff and French scientist, C. Le Bel in the same year (1874), independently argued that the spatial arrangement of four groups (valencies) around a central carbon is tetrahedral and if all the substituents attached to that carbon are different, the mirror image of the molecule is not superimposed (overlapped) on the molecule; such a carbon is called asymmetric carbon or stereocentre. The resulting molecule would lack symmetry and is referred to as asymmetric molecule. The asymmetry of the molecule along with non superimposability of mirror images is responsible for the optical activity in such organic compounds.
Jacobus Hendricus Van’t Hoff (1852-1911) received the first Nobel Prize in Chemistry in 1901 for his work on solutions.
objects: a sphere, a cube, a cone, are all identical to their mirror images and can be superimposed. However, many objects are non superimposable on their mirror images. For example, your left and right hand look similar but if you put your left hand on your right hand by moving them in the same plane, they do not coincide. The objects which are non- superimposable on their mirror image (like a pair of hands) are said to be chiral and this property is known as chirality. Chiral molecules are optically active, while the objects, which are, superimposable on their mirror images are called achiral. These molecules are optically inactive.The above test of molecular chirality can be applied to organic molecules by constructing models and its mirror images or by drawing three dimensional structures and attempting to superimpose them in our minds. There are other aids, however, that can assist us in recognisingchiral molecules. One such aid is the presence of a single asymmetric carbon atom. Let us consider two simple molecules propan-2-ol (Fig.10.5) and butan-2-ol
Fig 10.5: B is mirror image of A; B is rotated by 180o and C is obtained; C is superimposable on A.
(Fig.10.6) and their mirror images.
Fig 10.6: E is mirror image of D; E is rotated by 180o to get F and F is non superimposable on its mirror image D.
As you can see very clearly, propan-2-ol (A) does not contain an asymmetric carbon, as all the four groups attached to the tetrahedral carbon are not different. We rotate the mirror image (B) of the molecule by 180° (structure C) and try to overlap the structure (C) with the structure (A), these structures completely overlap. Thus propan-2-ol is an achiral molecule.
Butan-2-ol has four different groups attached to the tetrahedral carbon and as expected is chiral. Some common examples of chiral molecules such as 2-chlorobutane, 2, 3-dihyroxypropanal, (OHC–CHOH–CH2OH), bromochloro-iodomethane (BrClCHI), 2-bromopropanoic acid (H3C–CHBr–COOH), etc.
The stereoisomers related to each other as non- superimposable mirror images are called enantiomers (Fig. 10.7). A and B in Fig. 10.5 and D and E in Fig. 10.6 are enantiomers.
Fig. 10.7: A chiral molecule and its mirror image
Enantiomers possess identical physical properties namely, melting point, boiling point, refractive index, etc. They only differ with respect to the rotation of plane polarised light. If one of the enantiomer is dextro rotatory, the other will be laevo rotatory.
However, the sign of optical rotation is not necessarily related to the absolute (actual) configuration of the molecule.
A mixture containing two enantiomers in equal proportions will have zero optical rotation, as the rotation due to one isomer will be cancelled by the rotation due to the other isomer. Such a mixture is known as racemic mixture or racemic modification. A racemic mixture is represented by prefixing dl or (\(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGaeyySaelaaa!37E4! \pm \)) before the name, for example (\(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGaeyySaelaaa!37E4! \pm \)) butan-2-ol. The process of conversion of enantiomer into a racemic mixture is known as racemisation.
EXAMPLE 8
Identify chiral and achiral molecules in each of the following pair of compounds. (Wedge and Dash representations according to Class XI, Fig.12.1).
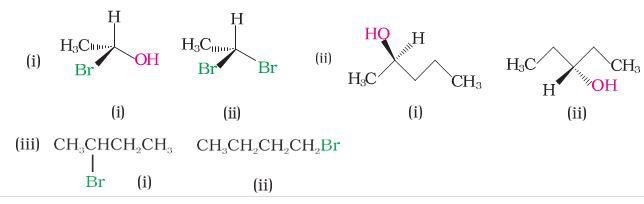
SOLUTION
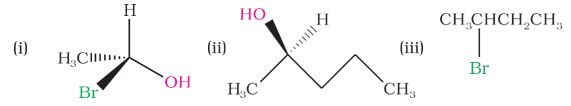
3. Retention: Retention of configuration is the preservation of the spatial arrangement of bonds to an asymmetric centre during a chemical reaction or transformation.
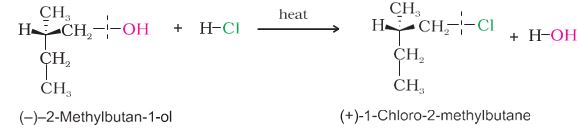
It is important to note that configuration at a symmetric centre in the reactant and product is same but the sign of optical rotation has changed in the product. This is so because two different compounds with same configuration at asymmetric centre may have different optical rotation. One may be dextrorotatory (plus sign of optical rotation) while other may be laevorotatory (negative sign of optical rotation).
4. Inversion, retention and racemisation: There are three outcomes for a reaction at an asymmetric carbon atom, when a bond directly linked to an asymmetric carbon atom is broken. Consider the replacement of a group X by Y in the following reaction;
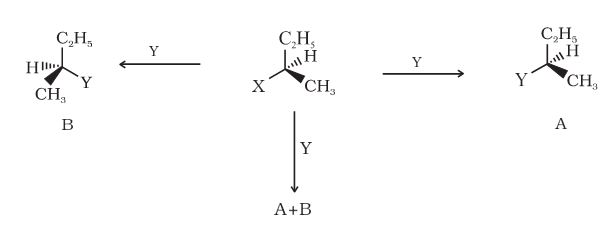
If (A) is the only compound obtained, the process is called retention of configuration. Note that configuration has been rotated in A. If (B) is the only compound obtained, the process is called inversion of configuration. Configuration has been inverted in B.
If a 50:50 mixture of A and B is obtained then the process is called racemisation and the product is optically inactive, as one isomer will rotate the plane polarised light in the direction opposite to another.
Now let us have a fresh look at SN1 and SN2 mechanisms by taking examples of optically active alkyl halides.
In case of optically active alkyl halides, the product formed as a result of SN2 mechanism has the inverted configuration as compared to the reactant. This is because the nucleophile attaches itself on the side opposite to the one where the halogen atom is present. When (–)-2-bromooctane is allowed to react with sodium hydroxide, (+)-octan-2-ol is formed with the –OH group occupying the position opposite to what bromide had occupied.
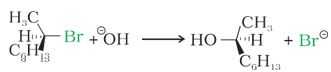
Thus, SN2 reactions of optically active halides are accompanied by inversion of configuration.
In case of optically active alkyl halides, SN1 reactions are accompanied by racemisation. Can you think of the reason why it happens? Actually the carbocation formed in the slow step being sp2 hybridised is planar (achiral). The attack of the nucleophile may be accomplished from either side of the plane of carbocation resulting in a mixture of products, one having the same configuration (the –OH attaching on the same position as halide ion) and the other having opposite configuration (the –OH attaching on the side opposite to halide ion). This may be illustrated by hydrolysis of optically active 2-bromobutane, which results in the formation of (\(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGaeyySaelaaa!37E4! \pm \))-butan-2-ol.
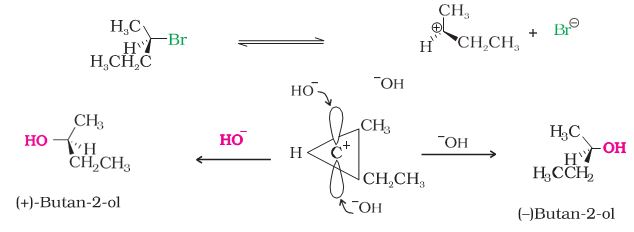
2. Elimination reactions
When a haloalkane with -hydrogen atom is heated with alcoholic solution of potassium hydroxide, there is elimination of hydrogen atom from \(\beta\)-carbon and a halogen atom from the \(\beta\)-carbon atom.

Location of \(\alpha\) and \(\beta\) carbon in a molecule Carbon on which halogen atom is directly attached is called \(\alpha\)-carbon and the carbon atom adjacent to this carbon is called \(\beta\)-carbon.

As a result, an alkene is formed as a product. Since \(\beta\)-hydrogen atom is involved in elimination, it is often called \(\beta\)-elimination.
If there is possibility of formation of more than one alkene due to the availability of more than one \(\beta\)-hydrogen atoms, usually one alkene is formed as the major product. These form part of a pattern first observed by Russian chemist, Alexander Zaitsev (also pronounced as Saytzeff) who in 1875 formulated a rule which can be summarised as “in dehydrohalogenation reactions, the preferred product is that alkene which has the greater number of alkyl groups attached to the doubly bonded carbon atoms.” Thus, 2-bromopentane gives pent-2-ene as the major product.
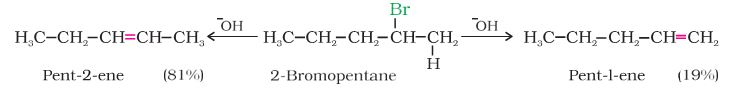
Elimination versus substitution
A chemical reaction is the result of competition; it is a race that is won by the fastest runner. A collection of molecules tend to do, by and large, what is easiest for them. An alkyl halide with \(\beta\)-hydrogen atoms when reacted with a base or a nucleophile has two competing routes: substitution (SN1 and SN2) and elimination. Which route will be taken up depends upon the nature of alkyl halide, strength and size of base/nucleophile and reaction conditions. Thus, a bulkier nucleophile will prefer to act as a base and abstracts a proton rather than approach a tetravalent carbon atom (steric reasons) and vice versa. Similarly, a primary alkyl halide will prefer a SN2 reaction, a secondary halide- SN2 or elimination depending upon the strength of base/nucleophile and a tertiary halide- SN1 or elimination depending upon the stability of carbocation or the more substituted alkene.
3. Reaction with metals
Most organic chlorides, bromides and iodides react with certain metals to give compounds containing carbon-metal bonds. Such compounds are known as organo-metallic compounds. An important class of organo-metallic compounds discovered by Victor Grignard in 1900 is alkyl magnesium halide, RMgX, referred as Grignard Reagents. These reagents are obtained by the reaction of haloalkanes with magnesium metal in dry ether.

Victor Grignard had a strange start in academic life for a chemist - he took a maths degree. When he eventually switched to chemistry, it was not to the mathematical province of physical chemistry but to organic chemistry. While attempting to find an efficient catalyst for the process of methylation, he noted that Zn in diethyl ether had been used for this purpose and wondered whether the Mg/ether combination might be successful. Grignard reagents were first reported in 1900 and Grignard used this work for his doctoral thesis in 1901. In 1910, Grignard obtained a professorship at the University of Nancy and in 1912, he was awarded the Nobel prize for Chemistry which he shared with Paul Sabatier who had made advances in nickel catalysed hydrogenation.
In the Grignard reagent, the carbon-magnesium bond is covalent but highly polar, with carbon pulling electrons from electropositive magnesium; the magnesium halogen bond is essentially ionic.
Grignard reagents are highly reactive and react with any source of proton to give hydrocarbons. Even water, alcohols, amines are sufficiently acidic to convert them to corresponding hydrocarbons.

It is therefore necessary to avoid even traces of moisture from a Grignard reagent. That is why reaction is carried out in dry ether. On the other hand, this could be considered as one of the methods for converting halides to hydrocarbons.
Wurtz reaction
Alkyl halides react with sodium in dry ether to give hydrocarbons containing double the number of carbon atoms present in the halide. This reaction is known as Wurtz reaction (Unit 13, Class XI).

Reactions OF Haloarenes
1. Nucleophilic substitution
Aryl halides are extremely less reactive towards nucleophilic substitution reactions due to the following reasons:
1. Resonance effect : In haloarenes, the electron pairs on halogen atom are in conjugation with \(\pi\)-electrons of the ring and the following resonating structures are possible.
C—Cl bond acquires a partial double bond character due to resonance. As a result, the bond cleavage in haloarene is difficult than haloalkane and therefore, they are less reactive towards nucleophilic substitution reaction.
2. Difference in hybridisation of carbon atom in C—X bond: In haloalkane, the carbon atom attached to halogen is sp3 hybridised while in case of haloarene, the carbon atom attached to halogen is sp2-hybridised.
The sp2 hybridised carbon with a greater s-character is more electronegative and can hold the electron pair of C—X bond more tightly than sp3-hybridised carbon in haloalkane with less s-chararcter. Thus, C—Cl bond length in haloalkane is 177pm while in haloarene is 169 pm. Since it is difficult to break a shorter bond than a longer bond, therefore, haloarenes are less reactive than haloalkanes towards nucleophilic substitution reaction.
3. Instability of phenyl cation: In case of haloarenes, the phenyl cation formed as a result of self-ionisation will not be stabilised by resonance and therefore, SN1 mechanism is ruled out.
4. Because of the possible repulsion, it is less likely for the electron rich nucleophile to approach electron rich arenes.
Replacement by hydroxyl group
Chlorobenzene can be converted into phenol by heating in aqueous sodium hydroxide solution at a temperature of 623K and a pressure of 300 atmospheres.
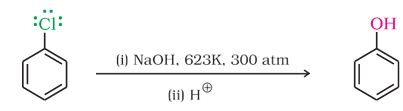
The presence of an electron withdrawing group (-NO2) at ortho- and para-positions increases the reactivity of haloarenes.

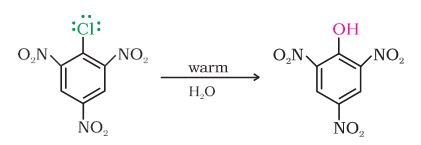
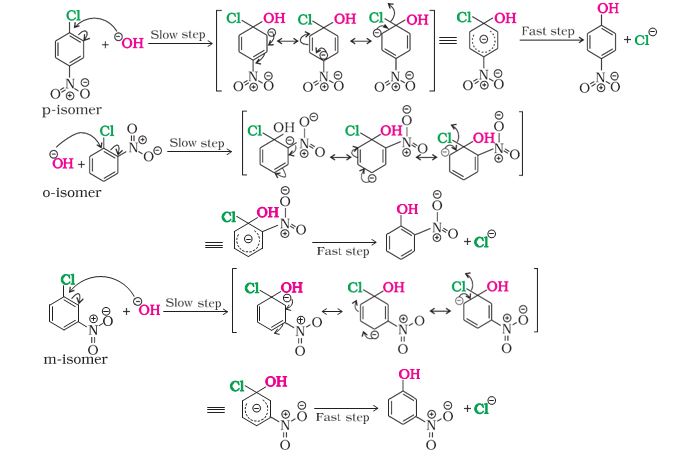
Can you think why does NO2 group show its effect only at ortho- and para- positions and not at meta- position?
As shown, the presence of nitro group at ortho- and para-positions withdraws the electron density from the benzene ring and thus facilitates the attack of the nucleophile on haloarene. The carbanion thus formed is stabilised through resonance. The negative charge appeared at ortho- and para- positions with respect to the halogen substituent is stabilised by –NO2 group while in case of meta-nitrobenzene, none of the resonating structures bear the negative charge on carbon atom bearing the –NO2 group. Therefore, the presence of nitro group at meta- position does not stabilise the negative charge and no effect on reactivity is observed by the presence of –NO2 group at meta-position.
2. Electrophilic substitution reactions
Haloarenes undergo the usual electrophilic reactions of the benzene ring such as halogenation, nitration, sulphonation and Friedel-Crafts reactions. Halogen atom besides being slightly deactivating is o, p- directing; therefore, further substitution occurs at ortho- and para- positions with respect to the halogen atom. The o, p-directing influence of halogen atom can be easily understood if we consider the resonating structures of halobenzene as shown:
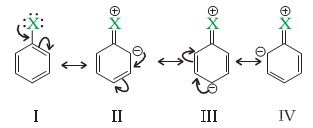
Due to resonance, the electron density increases more at ortho- and para-positions than at meta-positions. Further, the halogen atom because of its –I effect has some tendency to withdraw electrons from the benzene ring. As a result, the ring gets somewhat deactivated as compared to benzene and hence the electrophilic substitution reactions in haloarenes occur slowly and require more drastic conditions as compared to those in benzene.
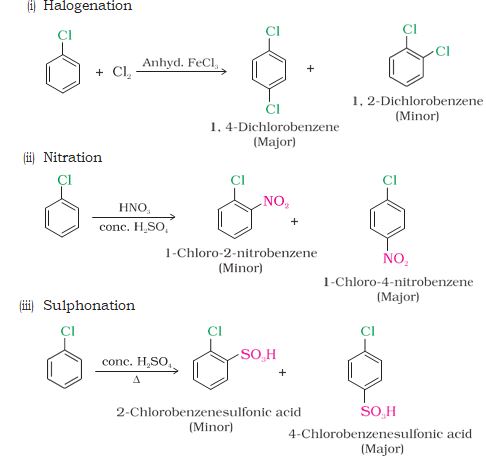
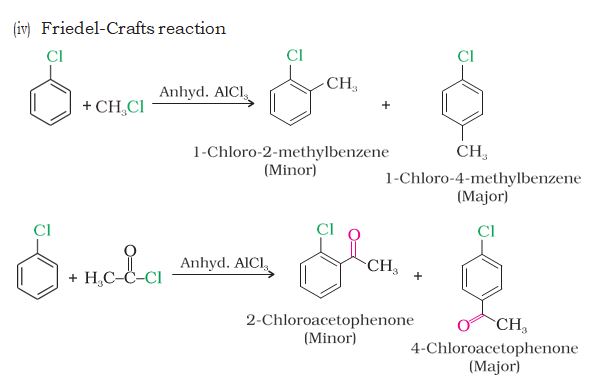
EXAMPLE 9
Although chlorine is an electron withdrawing group, yet it is ortho-, para- directing in electrophilic aromatic substitution reactions. Why?
SOLUTION
Chlorine withdraws electrons through inductive effect and releases electrons through resonance. Through inductive effect, chlorine destabilises the intermediate carbocation formed during the electrophilic substitution.
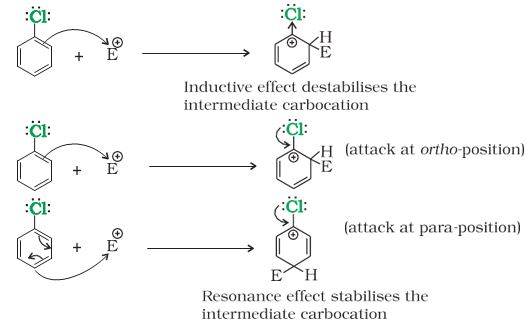
Through resonance, halogen tends to stabilise the carbocation and the effect is more pronounced at ortho- and para- positions. The inductive effect is stronger than resonance and causes net electron withdrawal and thus causes net deactivation. The resonance effect tends to oppose the inductive effect for the attack at ortho- and para- positions and hence makes the deactivation less for ortho- and para- attack. Reactivity is thus controlled by the stronger inductive effect and orientation is controlled by resonance effect.
3. Reaction with metals
Wurtz-Fittig reaction
A mixture of an alkyl halide and aryl halide gives an alkylarene when treated with sodium in dry ether and is called Wurtz-Fittig reaction.
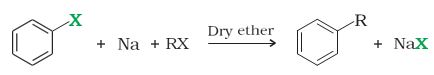
Fittig reaction
Aryl halides also give analogous compounds when treated with sodium in dry ether, in which two aryl groups are joined together. It is called Fittig reaction.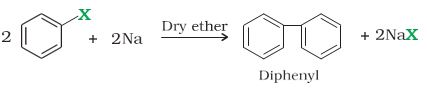
INTEXT QUESTIONS
Which alkyl halide from the following pairs would you expect to react more rapidly by an SN2 mechanism? Explain your answer.
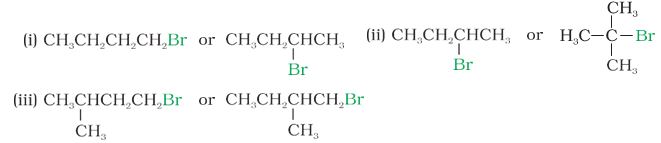
In the following pairs of halogen compounds, which compound undergoes faster SN1 reaction?

Identify A, B, C, D, E, R and R1 in the following:
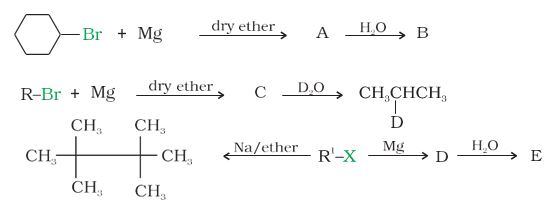
Polyhalogen Compounds
Carbon compounds containing more than one halogen atom are usually referred to as polyhalogen compounds. Many of these compounds are useful in industry and agriculture. Some polyhalogen compounds are described in this section.
DichlorO-methane (Methylene chloride)
Dichloromethane is widely used as a solvent as a paint remover, as a propellant in aerosols, and as a process solvent in the manufacture of drugs. It is also used as a metal cleaning and finishing solvent. Methylene chloride harms the human central nervous system. Exposure to lower levels of methylene chloride in air can lead to slightly impaired hearing and vision. Higher levels of methylene chloride in air cause dizziness, nausea, tingling and numbness in the fingers and toes. In humans, direct skin contact with methylene chloride causes intense burning and mild redness of the skin. Direct contact with the eyes can burn the cornea.
Trichloro-methane (Chloroform)
Chemically, chloroform is employed as a solvent for fats, alkaloids, iodine and other substances. The major use of chloroform today is in the production of the freon refrigerant R-22. It was once used as a general anaesthetic in surgery but has been replaced by less toxic, safer anaesthetics, such as ether. As might be expected from its use as an anaesthetic, inhaling chloroform vapours depresses the central nervous system. Breathing about 900 parts of chloroform per million parts of air (900 parts per million) for a short time can cause dizziness, fatigue, and headache. Chronic chloroform exposure may cause damage to the liver (where chloroform is metabolised to phosgene) and to the kidneys, and some people develop sores when the skin is immersed in chloroform. Chloroform is slowly oxidised by air in the presence of light to an extremely poisonous gas, carbonyl chloride, also known as phosgene. It is therefore stored in closed dark coloured bottles completely filled so that air is kept out.

Triiodo- methane (Iodoform)
It was used earlier as an antiseptic but the antiseptic properties are due to the liberation of free iodine and not due to iodoform itself. Due to its objectionable smell, it has been replaced by other formulations containing iodine.
Tetrachlo-romethane (Carbon tetrachloride)
It is produced in large quantities for use in the manufacture of refrigerants and propellants for aerosol cans. It is also used as feedstock in the synthesis of chlorofluorocarbons and other chemicals, pharmaceutical manufacturing, and general solvent use. Until the mid 1960s, it was also widely used as a cleaning fluid, both in industry, as a degreasing agent, and in the home, as a spot remover and as fire extinguisher. There is some evidence that exposure to carbon tetrachloride causes liver cancer in humans. The most common effects are dizziness, light headedness, nausea and vomiting, which can cause permanent damage to nerve cells. In severe cases, these effects can lead rapidly to stupor, coma, unconsciousness or death. Exposure to CCl4 can make the heart beat irregularly or stop. The chemical may irritate the eyes on contact. When carbon tetrachloride is released into the air, it rises to the atmosphere and depletes the ozone layer. Depletion of the ozone layer is believed to increase human exposure to ultraviolet rays, leading to increased skin cancer, eye diseases and disorders, and possible disruption of the immune system.
Freons
The chlorofluorocarbon compounds of methane and ethane are collectively known as freons. They are extremely stable, unreactive, non-toxic, non- corrosive and easily liquefiable gases. Freon 12 (CCl2F2) is one of the most common freons in industrial use. It is manufactured from tetrachloromethane by Swarts reaction. These are usually produced for aerosol propellants, refrigeration and air conditioning purposes. By 1974, total freon production in the world was about 2 billion pounds annually. Most freon, even that used in refrigeration, eventually makes its way into the atmosphere where it diffuses unchanged into the stratosphere. In stratosphere, freon is able to initiate radical chain reactions that can upset the natural ozone balance (Unit 14, Class XI).
p,,p’-Dichlo- rodiphenyl- trichloro- ethane(DDT)
DDT, the first chlorinated organic insecticides, was originally prepared in 1873, but it was not until 1939 that Paul Muller of Geigy Pharmaceuticals in Switzerland discovered the effectiveness of DDT as an insecticide. Paul Muller was awarded the Nobel Prize in Medicine and Physiology in 1948 for this discovery. The use of DDT increased enormously on a worldwide basis after World War II, primarily because of its effectiveness against the mosquito that spreads malaria and lice that carry typhus. However, problems related to extensive use of DDT began to appear in the late 1940s. Many species of insects developed resistance to DDT, and it was also discovered to have a high toxicity towards fish. The chemical stability of DDT and its fat solubility compounded the problem. DDT is not metabolised very rapidly by animals; instead, it is deposited and stored in the fatty tissues. If ingestion continues at a steady rate, DDT builds up within the animal over time. The use of DDT was banned in the United States in 1973, although it is still in use in some other parts of the world.
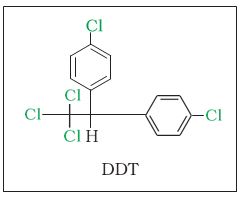
-
Alcohols, Phenols and Ethers.11
You have learnt that substitution of one or more hydrogen atom(s) from a hydrocarbon by another atom or a group of atoms result in the formation of an entirely new compound having altogether different properties and applications. Alcohols and phenols are formed when a hydrogen atom in a hydrocarbon, aliphatic and aromatic respectively, is replaced by –OH group. These classes of compounds find wide applications in industry as well as in day-to-day life. For instance, have you ever noticed that ordinary spirit used for polishing wooden furniture is chiefly a compound containing hydroxyl group, ethanol. The sugar we eat, the cotton used for fabrics, the paper we use for writing, are all made up of compounds containing –OH groups. Just think of life without paper; no note-books, books, news- papers, currency notes, cheques, certificates, etc. The magazines carrying beautiful photographs and interesting stories would disappear from our life. It would have been really a different world.
An alcohol contains one or more hydroxyl (OH) group(s) directly attached to carbon atom(s), of an aliphatic system (CH3OH) while a phenol contains –OH group(s) directly attached to carbon atom(s) of an aromatic system (C6H5OH).
The substitution of a hydrogen atom in a hydrocarbon by an alkoxy or aryloxy group (R–O/Ar–O) yields another class of compounds known as ‘ethers’, for example, CH3OCH3 (dimethyl ether). You may also visualise ethers as compounds formed by substituting the hydrogen atom of hydroxyl group of an alcohol or phenol by an alkyl or aryl group.
In this unit, we shall discuss the chemistry of three classes of compounds, namely — alcohols, phenols and ethers.
Classification
The classification of compounds makes their study systematic and hence simpler. Therefore, let us first learn how are alcohols, phenols and ethers classified?
Alcohols-Mono, Di, Tri or Polyhydric alcohols
Alcohols and phenols may be classified as mono–, di–, tri- or polyhydric compounds depending on whether they contain one, two, three or many hydroxyl groups respectively in their structures as given below:
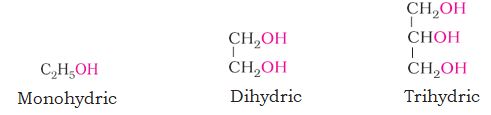
Monohydric alcohols may be further classified according to the hybridisation of the carbon atom to which the hydroxyl group is attached.
(1) Compounds containing Csp3 - OH bond: In this class of alcohols, the –OH group is attached to an sp3 hybridised carbon atom of an alkyl group. They are further classified as follows:
Primary, secondary and tertiary alcohols: In these three types of alcohols, the –OH group is attached to primary, secondary and tertiary carbon atom, respectively as depicted below:

Allylic alcohols: In these alcohols, the —OH group is attached to a sp3 hybridised carbon adjacent to the carbon-carbon double bond, that is to an allylic carbon. For example
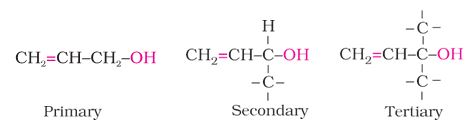
Benzylic alcohols: In these alcohols, the —OH group is attached to a sp3—hybridised carbon atom next to an aromatic ring. For example.
Allylic and benzylic alcohols may be primary, secondary or tertiary.
Compounds containing Csp2 - OH bond: These alcohols contain —OH group bonded to a carbon-carbon double bond, i.e., to a vinylic carbon or to an aryl carbon. These alcohols are also known as vinylic alcohols.Vinylic alcohol: CH2 = CH – OH
Phenols-Mono, Di and trihydric phenols
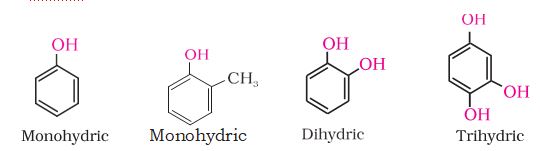
Ethers
Ethers are classified as simple or symmetrical, if the alkyl or aryl groups attached to the oxygen atom are the same, and mixed or unsymmetrical, if the two groups are different. Diethyl ether, C2H5OC2H5, is a symmetrical ether whereas C2H5OCH3 and C2H5OC6H5 are unsymmetrical ethers.
INTEXT QUESTION
Classify the following as primary, secondary and tertiary alcohols:

Identify allylic alcohols in the above examples.
Nomenclature
(a) Alcohols: The common name of an alcohol is derived from the common name of the alkyl group and adding the word alcohol to it. For example, CH3OH is methyl alcohol.
According to IUPAC system (Unit 12, Class XI), the name of an alcohol is derived from the name of the alkane from which the alcohol is derived, by substituting ‘e’ of alkane with the suffix ‘ol’. The position of substituents are indicated by numerals. For this, the longest carbon chain (parent chain) is numbered starting at the end nearest to the hydroxyl group. The positions of the –OH group and other substituents are indicated by using the numbers of carbon atoms to which these are attached. For naming polyhydric alcohols, the ‘e’ of alkane is retained and the ending ‘ol’ is added. The number of –OH groups is indicated by adding the multiplicative prefix, di, tri, etc., before ‘ol’. The positions of
–OH groups are indicated by appropriate locants, e.g., HO–CH2–CH2–OH is named as ethane–1, 2-diol. Table 11.1 gives common and IUPAC names of a few alcohols as examples.
Table 11.1: Common and IUPAC Names of Some Alcohols
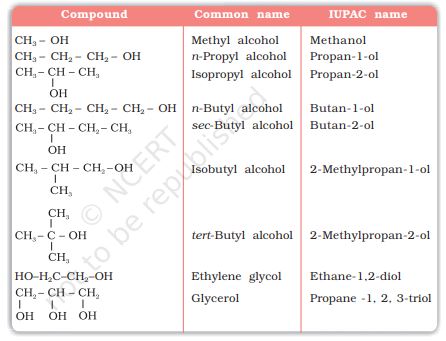
Cyclic alcohols are named using the prefix cyclo and considering the —OH group attached to C–1.
(b) Phenols: The simplest hydroxy derivative of benzene is phenol. It is its common name and also an accepted IUPAC name. As structure of phenol involves a benzene ring, in its substituted compounds the terms ortho (1,2- disubstituted), meta (1,3-disubstituted) and para (1,4-disubstituted) are often used in the common names.
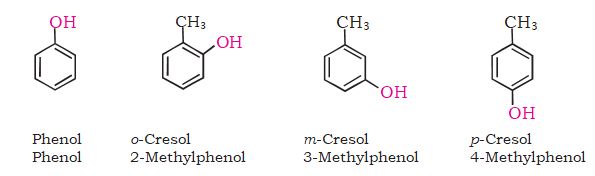
Common NAME IUPAC name
Dihydroxy derivatives of benzene are known as 1, 2-, 1, 3- and 1, 4-benzenediol.
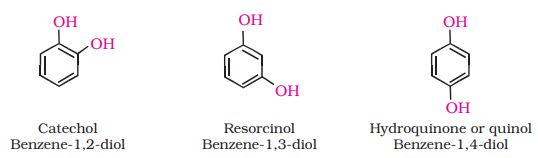
(c) Ethers: Common names of ethers are derived from the names of alkyl/ aryl groups written as separate words in alphabetical order and adding the word ‘ether’ at the end. For example, CH3OC2H5 is ethylmethyl ether.
If both the alkyl groups are the same, the prefix ‘di’ is added before the alkyl group. For example, C2H5OC2H5 is diethyl ether.
According to IUPAC system of nomenclature, ethers are regarded as hydrocarbon derivatives in which a hydrogen atom is replaced by an
–OR or –OAr group, where R and Ar represent alkyl and aryl groups, respectively. The larger (R) group is chosen as the parent hydrocarbon. The names of a few ethers are given as examples in Table 11.2.
Table 11.2: Common and IUPAC Names of Some Ethers
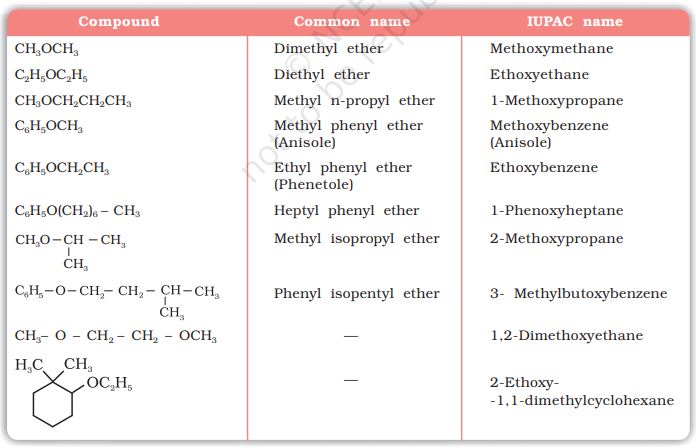
EXAMPLE 11
Give IUPAC names of the following compounds:
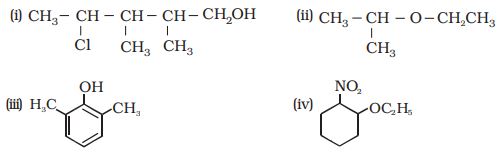
SOLUTION
(i) 4-Chloro-2,3-dimethylpentan-1-ol (ii) 2-Ethoxypropane
(iii) 2,6-Dimethylphenol (iv) 1-Ethoxy-2-nitrocyclohexane
INTEXT QUESTION
Name the following compounds according to IUPAC system.
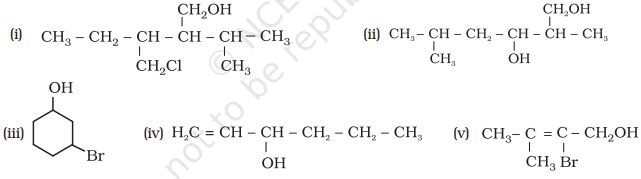
Structures OF FunctionalGroups
In alcohols, the oxygen of the –OH group is attached to carbon by a sigma (\(\sigma\)) bond formed by the overlap of a sp3 hybridised orbital of carbon with a sp3 hybridised orbital of oxygen. Fig. 11.1 depicts structural aspects of methanol, phenol and methoxymethane.
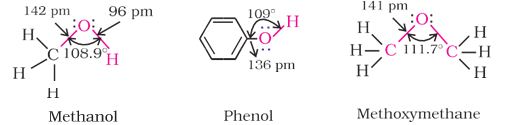
Fig. 11.1: Structures of methanol, phenol and methoxymethane
The bond angle
 in alcohols is slightly less than the tetrahedral angle (109°-2\(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGabGioayaafa % aaaa!36C4! 8'\)). It is due to the repulsion between the unshared electron pairs of oxygen. In phenols, the –OH group is attached to sp2 hybridised carbon of an aromatic ring. The carbon– oxygen bond length (136 pm) in phenol is slightly less than that in methanol. This is due to (i) partial double bond character on account of the conjugation of unshared electron pair of oxygen with the aromatic ring (Section 11.4.4) and (ii) sp2 hybridised state of carbon to which oxygen is attached.
in alcohols is slightly less than the tetrahedral angle (109°-2\(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGabGioayaafa % aaaa!36C4! 8'\)). It is due to the repulsion between the unshared electron pairs of oxygen. In phenols, the –OH group is attached to sp2 hybridised carbon of an aromatic ring. The carbon– oxygen bond length (136 pm) in phenol is slightly less than that in methanol. This is due to (i) partial double bond character on account of the conjugation of unshared electron pair of oxygen with the aromatic ring (Section 11.4.4) and (ii) sp2 hybridised state of carbon to which oxygen is attached.In ethers, the four electron pairs, i.e., the two bond pairs and two lone pairs of electrons on oxygen are arranged approximately in a tetrahedral arrangement. The bond angle is slightly greater than the tetrahedral angle due to the repulsive interaction between the two bulky (–R) groups. The C–O bond length (141 pm) is almost the same as in alcohols.
Alcohols anD Phenols
Preparation of Alcohols
Alcohols are prepared by the following methods :
1.From alkenes
(1) By acid catalysed hydration: Alkenes react with water in the presence of acid as catalyst to form alcohols. In case of unsymmetrical alkenes, the addition reaction takes place in accordance with Markovnikov’s rule (Unit 13, Class XI).
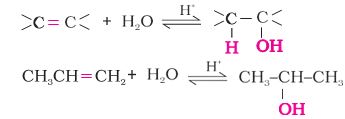
Mechanism
The mechanism of the reaction involves the following three steps:
Step 1: Protonation of alkene to form carbocation by electrophilic attack of H3O+.

Step 2: Nucleophilic attack of water on carbocation.
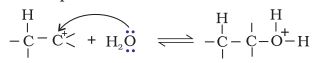
Step 3: Deprotonation to form an alcohol.

(2) By hydroboration–oxidation: Diborane (BH3)2 reacts with alkenes to give trialkyl boranes as addition product. This is oxidised to alcohol by hydrogen peroxide in the presence of aqueous sodium hydroxide.

The addition of borane to the double bond takes place in such a manner that the boron atom gets attached to the sp2 carbon carrying greater number of hydrogen atoms. The alcohol so formed looks as if it has been formed by the addition of water to the alkene in a way opposite to the Markovnikov’s rule. In this reaction, alcohol is obtained in excellent yield.
Hydroboration - oxidation was first reported by H.C. Brown in 1959. For his studies on boron containing organic compounds, Brown shared the 1979 Nobel prize in Chemistry with G. Wittig.
2. From carbonyl compounds
(1) By reduction of aldehydes and ketones: Aldehydes and ketones are reduced to the corresponding alcohols by addition of hydrogen in the presence of catalysts (catalytic hydrogenation). The usual catalyst is a finely divided metal such as platinum, palladium or nickel. It is also prepared by treating aldehydes and ketones with sodium borohydride (NaBH4) or lithium aluminium hydride (LiAlH4). Aldehydes yield primary alcohols whereas ketones give secondary alcohols.
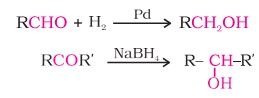
(2) By reduction of carboxylic acids and esters: Carboxylic acids are reduced to primary alcohols in excellent yields by lithium aluminium hydride, a strong reducing agent.
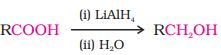
However, LiAlH4 is an expensive reagent, and therefore, used for preparing special chemicals only. Commercially, acids are reduced to alcohols by converting them to the esters (Section 11.4.4), followed by their reduction using hydrogen in the presence of catalyst (catalytic hydrogenation).

The numbers in front of the reagents along the arrow indicate that the second reagent is added only when the reaction with first is complete.
3. From Grignard reagents
Alcohols are produced by the reaction of Grignard reagents (Unit 10, Class XII) with aldehydes and ketones.
The first step of the reaction is the nucleophilic addition of Grignard reagent to the carbonyl group to form an adduct. Hydrolysis of the adduct yields an alcohol.

The overall reactions using different aldehydes and ketones are as follows:
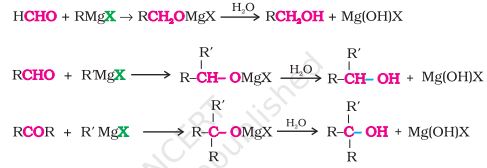
You will notice that the reaction produces a primary alcohol with methanal, a secondary alcohol with other aldehydes and tertiary alcohol with ketones.
The reaction of Grignard reagents with methanal produces a primary alcohol, with other aldehydes, secondary alcohols and with ketones, tertiary alcohols.
EXAMPLE 12
Give the structures and IUPAC names of the products expected from the following reactions:
a.Catalytic reduction of butanal.
b. Hydration of propene in the presence of dilute sulphuric acid.
c. Reaction of propanone with methylmagnesium bromide followed by hydrolysis.
SOLUTION
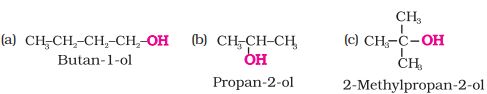
Preparation of Phenols
Phenol, also known as carbolic acid, was first isolated in the early nineteenth century from coal tar. Nowadays, phenol is commercially produced synthetically. In the laboratory, phenols are prepared from benzene derivatives by any of the following methods:
1. From haloarenes
Chlorobenzene is fused with NaOH at 623K and 320 atmospheric pressure. Phenol is obtained by acidification of sodium phenoxide so produced (Unit 10, Class XII).
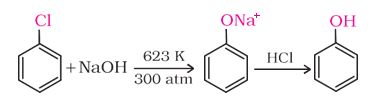
2. From benzenesulphonic acid
Benzene is sulphonated with oleum and benzene sulphonic acid so formed is converted to sodium phenoxide on heating with molten sodium hydroxide. Acidification of the sodium salt gives phenol.
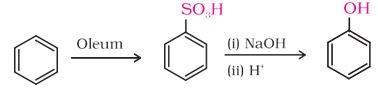
3. From diazonium salts
A diazonium salt is formed by treating an aromatic primary amine with nitrous acid (NaNO2 + HCl) at 273-278 K. Diazonium salts are hydrolysed to phenols by warming with water or by treating with dilute acids (Unit 13, Class XII).
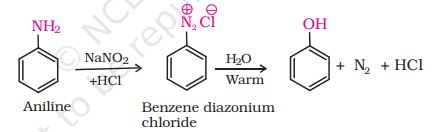
Most of the worldwide production of phenol is from cumene.
4. From cumene
Phenol is manufactured from the hydrocarbon, cumene. Cumene (isopropylbenzene) is oxidised in the presence of air to cumene hydroperoxide. It is converted to phenol and acetone by treating it with dilute acid. Acetone, a by-product of this reaction, is also obtained in large quantities by this method.
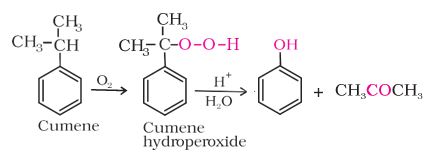
INTEXT QUESTION
Show how are the following alcohols prepared by the reaction of a suitable Grignard reagent on methanal ?

Write structures of the products of the following reactions:
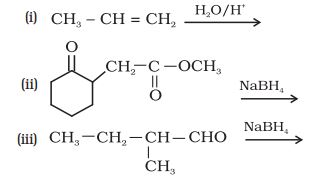
Physical Properties
Alcohols and phenols consist of two parts, an alkyl/aryl group and a hydroxyl group. The properties of alcohols and phenols are chiefly due to the hydroxyl group. The nature of alkyl and aryl groups simply modify these properties.
Boiling Points
The boiling points of alcohols and phenols increase with increase in the number of carbon atoms (increase in van der Waals forces). In alcohols, the boiling points decrease with increase of branching in carbon chain (because of decrease in van der Waals forces with decrease in surface area).
The –OH group in alcohols and phenols is involved in intermolecular hydrogen bonding as shown below:
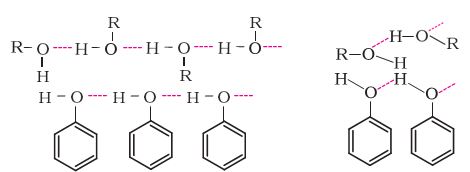
It is interesting to note that boiling points of alcohols and phenols are higher in comparison to other classes of compounds, namely hydrocarbons, ethers, haloalkanes and haloarenes of comparable molecular masses. For example, ethanol and propane have comparable molecular masses but their boiling points differ widely. The boiling point of methoxymethane is intermediate of the two boiling points.
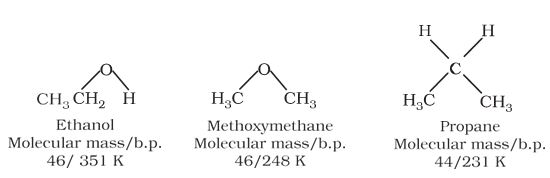
The high boiling points of alcohols are mainly due to the presence of intermolecular hydrogen bonding in them which is lacking in ethers and hydrocarbons.
Solubility
Solubility of alcohols and phenols in water is due to their ability to form hydrogen bonds with water molecules as shown. The solubility decreases with increase in size of alkyl/aryl (hydro- phobic) groups. Several of the lower molecular mass alcohols are miscible with water in all proportions.
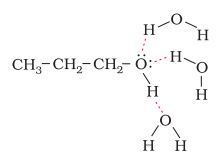
EXAMPLE 13
Arrange the following sets of compounds in order of their increasing boiling points:
a. Pentan-1-ol, butan-1-ol, butan-2-ol, ethanol, propan-1-ol, methanol.
b. Pentan-1-ol, n-butane, pentanal, ethoxyethane.
SOLUTION
(a) Methanol, ethanol, propan-1-ol, butan-2-ol, butan-1-ol, pentan-1-ol.
(b) n-Butane, ethoxyethane, pentanal and pentan-1-ol.
Chemical Reactions
Alcohols are versatile compounds. They react both as nucleophiles and electrophiles. The bond between O–H is broken when alcohols react as nucleophiles.
Alcohols as nucleophiles
(1)
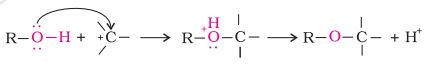
(2) The bond between C–O is broken when they react as electrophiles. Protonated alcohols react in this manner.
Protonated alcohols as electrophiles

Based on the cleavage of O–H and C–O bonds, the reactions of alcohols and phenols may be divided into two groups:
(a) Reactions involving cleavage of O–H bond
1. Acidity of alcohols and phenols
(1) Reaction with metals: Alcohols and phenols react with active metals such as sodium, potassium and aluminium to yield corresponding alkoxides/phenoxides and hydrogen.
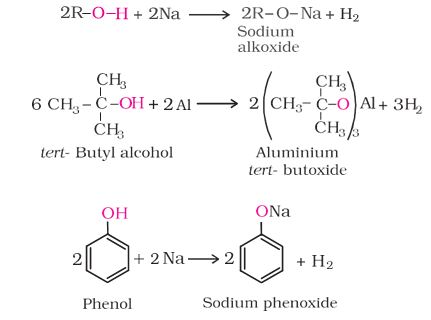
In addition to this, phenols react with aqueous sodium hydroxide to form sodium phenoxides.
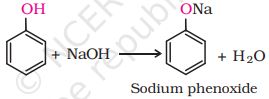
The above reactions show that alcohols and phenols are acidic in nature. In fact, alcohols and phenols are Brönsted acids i.e., they can donate a proton to a stronger base (B:).
(2) Acidity of alcohols: The acidic character of alcohols is due to the polar nature of O–H bond. An electron-releasing group (–CH3, –C2H5) increases electron density on oxygen tending to decrease the polarity of O-H bond. This decreases the acid strength. For this reason, the acid strength of alcohols decreases in the following order:
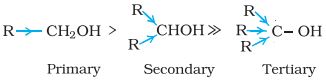
Alcohols are, however, weaker acids than water. This can be illustrated by the reaction of water with an alkoxide.
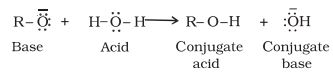
This reaction shows that water is a better proton donor (i.e., stronger acid) than alcohol. Also, in the above reaction, we note that an alkoxide ion is a better proton acceptor than hydroxide ion, which suggests that alkoxides are stronger bases (sodium ethoxide is a stronger base than sodium hydroxide).
Alcohols act as Bronsted bases as well. It is due to the presence of unshared electron pairs on oxygen, which makes them proton acceptors.
(3) Acidity of phenols: The reactions of phenol with metals (e.g., sodium, aluminium) and sodium hydroxide indicate its acidic nature. The hydroxyl group, in phenol is directly attached to the sp2 hybridised carbon of benzene ring which acts as an electron withdrawing group. Due to this, the charge distribution in phenol molecule, as depicted in its resonance structures, causes the oxygen of –OH group to be positive.
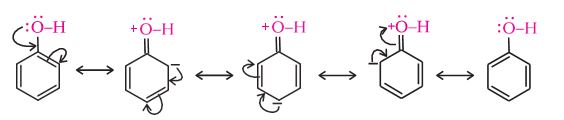
The reaction of phenol with aqueous sodium hydroxide indicates that phenols are stronger acids than alcohols and water. Let us examine how a compound in which hydroxyl group attached to an aromatic ring is more acidic than the one in which hydroxyl group is attached to an alkyl group.
The ionisation of an alcohol and a phenol takes place as follows:
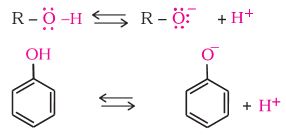
Due to the higher electronegativity of sp2 hybridised carbon of phenol to which –OH is attached, electron density decreases on oxygen. This increases the polarity of O–H bond and results in an increase in ionisation of phenols than that of alcohols. Now let us examine the stabilities of alkoxide and phenoxide ions. In alkoxide ion, the negative charge is localised on oxygen while in phenoxide ion, the charge is delocalised. The delocalisation of negative charge (structures I-V) makes phenoxide ion more stable and favours the ionisation of phenol. Although there is also charge delocalisation in phenol, its resonance structures have charge separation due to which the phenol molecule is less stable than phenoxide ion.
In substituted phenols, the presence of electron withdrawing groups such as nitro group, enhances the acidic strength of phenol. This effect is more pronounced when such a group is present at ortho and para positions. It is due to the effective delocalisation of negative charge in phenoxide ion when substituent is at ortho or para position. On the other hand, electron releasing groups, such as alkyl groups, in general, do not favour the formation of phenoxide ion resulting in decrease in acid strength. Cresols, for example, are less acidic than phenol.
The greater the pKa value, the weaker the acid.
Table 11.3: pKa Values of some Phenols and Ethanol
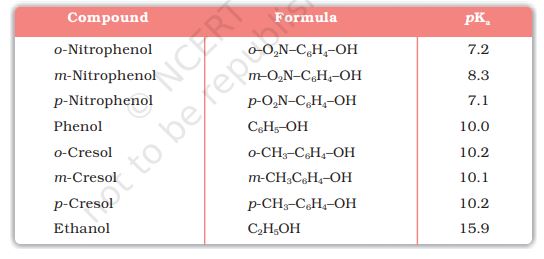
From the above data, you will note that phenol is million times more acidic than ethanol.
EXAMPLE 4
Arrange the following compounds in increasing order of their acid strength: Propan-1-ol, 2,4,6-trinitrophenol, 3-nitrophenol, 3,5-dinitrophenol, phenol, 4-methylphenol.
SOLUTION
Propan-1-ol, 4-methylphenol, phenol, 3-nitrophenol, 3,5-dinitrophenol, 2,4, 6-trinitrophenol.
2. Esterification
Alcohols and phenols react with carboxylic acids, acid chlorides and acid anhydrides to form esters.
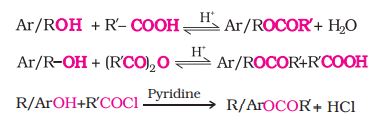
Aspirin possesses analgesic, anti- inflammatory and antipyretic properties.
The reaction with carboxylic acid and acid anhydride is carried out in the presence of a small amount of concentrated sulphuric acid. The reaction is reversible, and therefore, water is removed as soon as it is formed. The reaction with acid chloride is carried out in the presence of a base (pyridine) so as to neutralise HCl which is formed during the reaction. It shifts the equilibrium to the right hand side. The introduction of acetyl (CH3CO) group in alcohols or phenols is known as acetylation. Acetylation of salicylic acid produces aspirin.

(b) Reactions involving cleavage of carbon – oxygen (C–O) bond in alcohols
The reactions involving cleavage of C–O bond take place only in alcohols. Phenols show this type of reaction only with zinc.
1. Reaction with hydrogen halides: Alcohols react with hydrogen halides to form alkyl halides (Refer Unit 10, Class XII).
ROH + HX \(\to\) R–X + H2O
The difference in reactivity of three classes of alcohols with HCl distinguishes them from one another (Lucas test). Alcohols are soluble in Lucas reagent (conc. HCl and ZnCl2) while their halides are immiscible and produce turbidity in solution. In case of tertiary alcohols, turbidity is produced immediately as they form the halides easily. Primary alcohols do not produce turbidity at room temperature.
2. Reaction with phosphorus trihalides: Alcohols are converted to alkyl bromides by reaction with phosphorus tribromide (Refer Unit 10, Class XII).
3. Dehydration: Alcohols undergo dehydration (removal of a molecule of water) to form alkenes on treating with a protic acid e.g., concentrated H2SO4 or H3PO4, or catalysts such as anhydrous zinc chloride or alumina (Unit 13, Class XI).
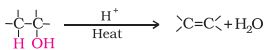
Ethanol undergoes dehydration by heating it with concentrated H2SO4 at 443 K.
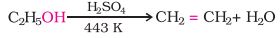
Secondary and tertiary alcohols are dehydrated under milder conditions. For example
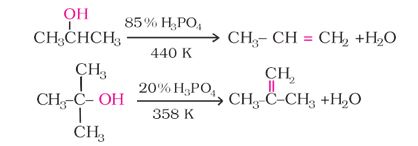
Thus, the relative ease of dehydration of alcohols follows the following order:
Tertiary > Secondary > Primary
The mechanism of dehydration of ethanol involves the following steps:
Mechanism
Step 1: Formation of protonated alcohol.
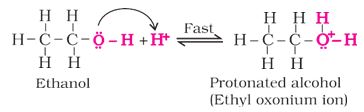
Step 2: Formation of carbocation: It is the slowest step and hence, the rate determining step of the reaction.
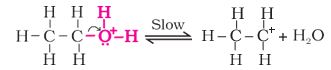
Step 3: Formation of ethene by elimination of a proton.
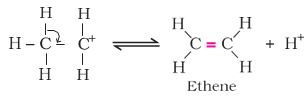
The acid used in step 1 is released in step 3. To drive the equilibrium to the right, ethene is removed as it is formed.
4. Oxidation: Oxidation of alcohols involves the formation of a carbon- oxygen double bond with cleavage of an O-H and C-H bonds.
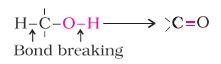
Such a cleavage and formation of bonds occur in oxidation reactions. These are also known as dehydrogenation reactions as these involve loss of dihydrogen from an alcohol molecule. Depending on the oxidising agent used, a primary alcohol is oxidised to an aldehyde which in turn is oxidised to a carboxylic acid.
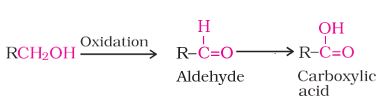
Strong oxidising agents such as acidified potassium permanganate are used for getting carboxylic acids from alcohols directly. CrO3 in anhydrous medium is used as the oxidising agent for the isolation of aldehydes.
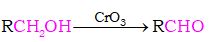
A better reagent for oxidation of primary alcohols to aldehydes in good yield is pyridinium chlorochromate (PCC), a complex of chromium trioxide with pyridine and HCl.

Secondary alcohols are oxidised to ketones by chromic anhyride (CrO3).
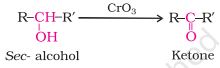
Tertiary alcohols do not undergo oxidation reaction. Under strong reaction conditions such as strong oxidising agents (KMnO4) and elevated temperatures, cleavage of various C-C bonds takes place and a mixture of carboxylic acids containing lesser number of carbon atoms is formed.
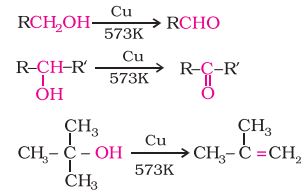
Biological oxidation of methanol and ethanol in the body produces the corresponding aldehyde followed by the acid. At times the alcoholics, by mistake, drink ethanol, mixed with methanol also called denatured alcohol. In the body, methanol is oxidised first to methanal and then to methanoic acid, which may cause blindness and death. A methanol poisoned patient is treated by giving intravenous infusions of diluted ethanol. The enzyme responsible for oxidation of aldehyde (HCHO) to acid is swamped allowing time for kidneys to excrete methanol.
(c) Reactions of phenols
Following reactions are shown by phenols only.
1. Electrophilic aromatic substitution
In phenols, the reactions that take place on the aromatic ring are electrophilic substitution reactions (Unit 13, Class XI). The –OH group attached to the benzene ring activates it towards electrophilic substitution. Also, it directs the incoming group to ortho and para positions in the ring as these positions become electron rich due to the resonance effect caused by –OH group. The resonance structures are shown under acidity of phenols.
Common electrophilic aromatic substitution reactions taking place in phenol are as follows:
(1) Nitration: With dilute nitric acid at low temperature (298 K), phenol yields a mixture of ortho and para nitrophenols.
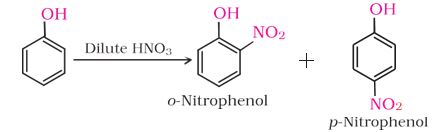
The ortho and para isomers can be separated by steam distillation. o-Nitrophenol is steam volatile due to intramolecular hydrogen bonding while p-nitrophenol is less volatile due to intermolecular hydrogen bonding which causes the association of molecules.
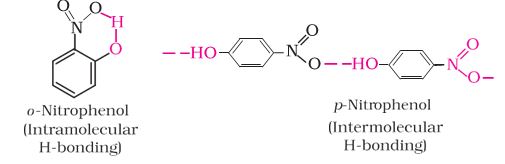
With concentrated nitric acid, phenol is converted to 2,4,6-trinitrophenol. The product is commonly known as picric acid. The yield of the reaction product is poor.
2, 4, 6 - Trinitrophenol is a strong acid due to the presence of three electron withdrawing –NO2 groups which facilitate the release of hydrogen ion.
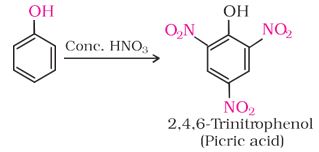
Nowadays picric acid is prepared by treating phenol first with concentrated sulphuric acid which converts it to phenol-2,4-disulphonic acid, and then with concentrated nitric acid to get 2,4,6-trinitrophenol. Can you write the equations of the reactions involved?
(2) Halogenation: On treating phenol with bromine, different reaction products are formed under different experimental conditions.
(a) When the reaction is carried out in solvents of low polarity such as CHCl3 or CS2 and at low temperature, monobromophenols are formed.
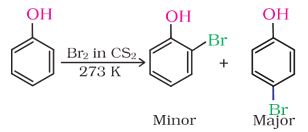
The usual halogenation of benzene takes place in the presence of a Lewis acid, such as FeBr3 (Unit 10, Class XII), which polarises the halogen molecule. In case of phenol, the polarisation of bromine molecule takes place even in the absence of Lewis acid. It is due to the highly activating effect of –OH group attached to the benzene ring.
(b) When phenol is treated with bromine water, 2,4,6-tribromophenol is formed as white precipitate.

EXAMPLE 5
Write the structures of the major products expected from the following reactions:
a. Mononitration of 3-methylphenol
b. Dinitration of 3-methylphenol
c. Mononitration of phenyl methanoate.
SOLUTION
The combined influence of –OH and –CH3 groups determine the position of the incoming group.
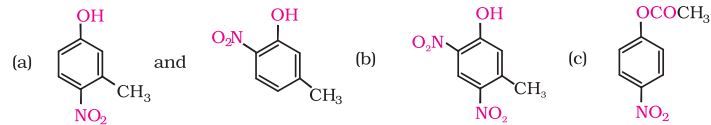
2. Kolbe’s reaction
Phenoxide ion generated by treating phenol with sodium hydroxide is even more reactive than phenol towards electrophilic aromatic substitution. Hence, it undergoes electrophilic substitution with carbon dioxide, a weak electrophile. Ortho hydroxybenzoic acid is formed as the main reaction product.
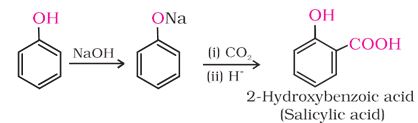
3. Reimer-Tiemann reaction
On treating phenol with chloroform in the presence of sodium hydroxide, a –CHO group is introduced at ortho position of benzene ring. This reaction is known as Reimer - Tiemann reaction.
The intermediate substituted benzal chloride is hydrolysed in the presence of alkali to produce salicylaldehyde.
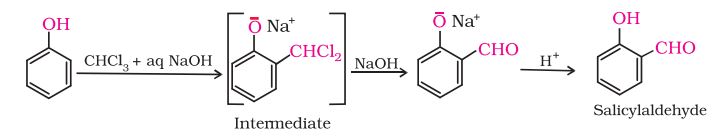
4. Reaction of phenol with zinc dust
Phenol is converted to benzene on heating with zinc dust.
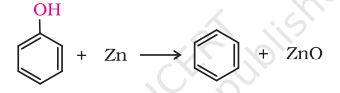
5. Oxidation
Oxidation of phenol with chromic acid produces a conjugated diketone known as benzoquinone. In the presence of air, phenols are slowly oxidised to dark coloured mixtures containing quinones.
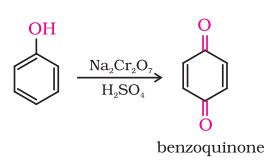
INTEXT QUESTION
Give structures of the products you would expect when each of the following alcohol reacts with (a) HCl –ZnCl2 (b) HBr and (c) SOCl2.
(i) Butan-1-ol (ii) 2-Methylbutan-2-ol
Predict the major product of acid catalysed dehydration of
(i) 1-methylcyclohexanol and (ii) butan-1-ol
Ortho and para nitrophenols are more acidic than phenol. Draw the resonance structures of the corresponding phenoxide ions.
Write the equations involved in the following reactions:
(i) Reimer - Tiemann reaction (ii) Kolbe’s reaction
SOME Commercially Important Alcohols
Methanol and ethanol are among the two commercially important alcohols.
1. Methanol
Methanol, CH3OH, also known as ‘wood spirit’, was produced by destructive distillation of wood. Today, most of the methanol is produced by catalytic hydrogenation of carbon monoxide at high pressure and temperature and in the presence of ZnO – Cr2O3 catalyst.

Methanol is a colourless liquid and boils at 337 K. It is highly poisonous in nature. Ingestion of even small quantities of methanol can cause blindness and large quantities causes even death. Methanol is used as a solvent in paints, varnishes and chiefly for making formaldehyde.
2.Ethanol
Ethanol, C2H5OH, is obtained commercially by fermentation, the oldest method is from sugars. The sugar in molasses, sugarcane or fruits such as grapes is converted to glucose and fructose, (both of which have the formula C6H12O6), in the presence of an enzyme, invertase. Glucose and fructose undergo fermentation in the presence of another enzyme, zymase, which is found in yeast.

In wine making, grapes are the source of sugars and yeast. As grapes ripen, the quantity of sugar increases and yeast grows on the outer skin. When grapes are crushed, sugar and the enzyme come in contact and fermentation starts. Fermentation takes place in anaerobic conditions i.e. in absence of air. Carbon dioxide is released during fermentation.
The action of zymase is inhibited once the percentage of alcohol formed exceeds 14 percent. If air gets into fermentation mixture, the oxygen of air oxidises ethanol to ethanoic acid which in turn destroys the taste of alcoholic drinks.
Ethanol is a colourless liquid with boiling point 351 K. It is used as a solvent in paint industry and in the preparation of a number of carbon compounds. The commercial alcohol is made unfit for drinking by mixing in it some copper sulphate (to give it a colour) and pyridine (a foul smelling liquid). It is known as denaturation of alcohol.
Nowadays, large quantities of ethanol are obtained by hydration of ethene (Section 11.4).
Ingestion of ethanol acts on the central nervous system. In moderate amounts, it affects judgment and lowers inhibitions. Higher concentrations cause nausea and loss of consciousness. Even at higher concentrations, it interferes with spontaneous respiration and can be fatal.
Ethers
Preparation of Ethers
1. By dehydration of alcohols
Alcohols undergo dehydration in the presence of protic acids (H2SO4, H3PO4). The formation of the reaction product, alkene or ether depends on the reaction conditions. For example, ethanol is dehydrated to ethene in the presence of sulphuric acid at 443 K. At 413 K, ethoxyethane is the main product.
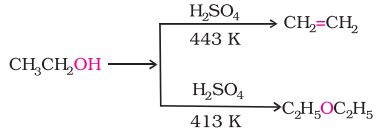
The formation of ether is a nucleophilic bimolecular reaction (SN2) involving the attack of alcohol molecule on a protonated alcohol, as indicated below:

Acidic dehydration of alcohols, to give an alkene is also associated with substitution reaction to give an ether.
Diethyl ether has been used widely as an inhalation anaesthetic. But due to its slow effect and an unpleasant recovery period, it has been replaced, as an anaesthetic, by other compounds.
The method is suitable for the preparation of ethers having primary alkyl groups only. The alkyl group should be unhindered and the temperature be kept low. Otherwise the reaction favours the formation of alkene. The reaction follows SN1 pathway when the alcohol is secondary or tertiary about which you will learn in higher classes. However, the dehydration of secondary and tertiary alcohols to give corresponding ethers is unsuccessful as elimination competes over substitution and as a consequence, alkenes are easily formed.
Can you explain why is bimolecular dehydration not appropriate for the preparation of ethyl methyl ether?
2. Williamson synthesis
It is an important laboratory method for the preparation of symmetrical and unsymmetrical ethers. In this method, an alkyl halide is allowed to react with sodium alkoxide.

Alexander William Williamson (1824–1904) was born in London of Scottish parents. In 1849, he became Professor of Chemistry at University College, London.
Ethers containing substituted alkyl groups (secondary or tertiary) may also be prepared by this method. The reaction involves SN2 attack of an alkoxide ion on primary alkyl halide.
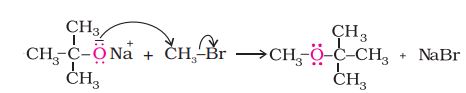
Better results are obtained if the alkyl halide is primary. In case of secondary and tertiary alkyl halides, elimination competes over substitution. If a tertiary alkyl halide is used, an alkene is the only reaction product and no ether is formed. For example, the reaction of CH3ONa with (CH3)3C–Br gives exclusively 2-methylpropene.

It is because alkoxides are not only nucleophiles but strong bases as well. They react with alkyl halides leading to elimination reaction.
EXAMPLE 6
The following is not an appropriate reaction for the preparation of t-butyl ethyl ether.
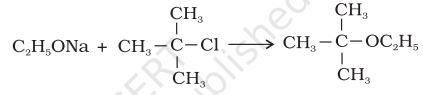
1. What would be the major product of this reaction ?
2. Write a suitable reaction for the preparation of t-butylethyl ether.
SOLUTION
1. The major product of the given reaction is 2-methylprop-1-ene. It is because sodium ethoxide is a strong nucleophile as well as a strong base. Thus elimination reaction predominates over substitution.
2.
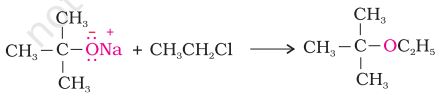
Phenols are also converted to ethers by this method. In this, phenol is used as the phenoxide moiety.
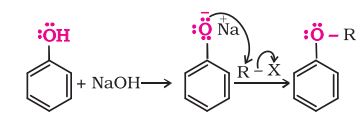
Physical Properties
The C-O bonds in ethers are polar and thus, ethers have a net dipole moment. The weak polarity of ethers do not appreciably affect their boiling points which are comparable to those of the alkanes of comparable molecular masses but are much lower than the boiling points of alcohols as shown in the following cases:

The large difference in boiling points of alcohols and ethers is due to the presence of hydrogen bonding in alcohols.
The miscibility of ethers with water resembles those of alcohols of the same molecular mass. Both ethoxyethane and butan-1-ol are miscible to almost the same extent i.e., 7.5 and 9 g per 100 mL water, respectively while pentane is essentially immiscible with water. Can you explain this observation ? This is due to the fact that just like alcohols, oxygen of ether can also form hydrogen bonds with water molecule as shown:
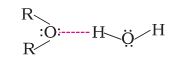
Chemical Reactions
1. Cleavage of C–O bond in ethers
Ethers are the least reactive of the functional groups. The cleavage of C-O bond in ethers takes place under drastic conditions with excess of hydrogen halides. The reaction of dialkyl ether gives two alkyl halide molecules.
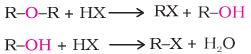
Alkyl aryl ethers are cleaved at the alkyl-oxygen bond due to the more stable aryl-oxygen bond. The reaction yields phenol and alkyl halide.
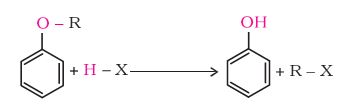
Ethers with two different alkyl groups are also cleaved in the same manner.
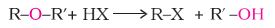
The order of reactivity of hydrogen halides is as follows: HI > HBr > HCl. The cleavage of ethers takes place with concentrated HI or HBr at high temperature.
Mechanism
The reaction of an ether with concentrated HI starts with protonation of ether molecule. Step 1:
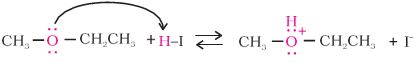
The reaction takes place with HBr or HI because these reagents are sufficiently acidic.
Step 2:
Iodide is a good nucleophile. It attacks the least substituted carbon of the oxonium ion formed in step 1 and displaces an alcohol molecule by SN2 mechanism.
Thus, in the cleavage of mixed ethers with two different alkyl groups, the alcohol and alkyl iodide formed, depend on the nature of alkyl groups. When primary or secondary alkyl groups are present, it is the lower alkyl group that forms alkyl iodide (SN2 reaction).
Therefore the attack by I– ion breaks O–CH3 bond to form CH3I. Phenols do not react further to give halides because the sp2 hybridised carbon of phenol cannot undergo nucleophilic substitution reaction needed for conversion to the halide.
EXAMPLE 7
Give the major products that are formed by heating each of the following ethers with HI.
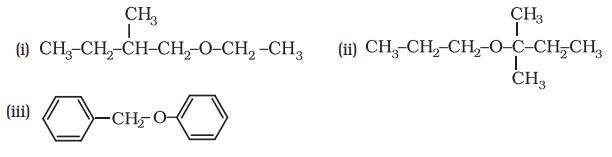
SOLUTION
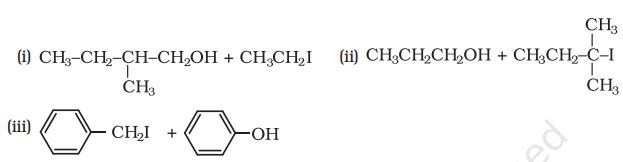
2. Electrophilic substitution
The alkoxy group (-OR) is ortho, para directing and activates the aromatic ring towards electrophilic substitution in the same way as in phenol.
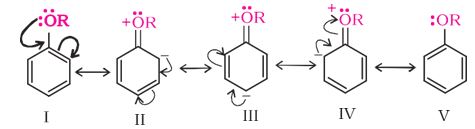
(1). Halogenation: Phenylalkyl ethers undergo usual halogenation in the benzene ring, e.g., anisole undergoes bromination with bromine in ethanoic acid even in the absence of iron (III) bromide catalyst. It is due to the activation of benzene ring by the methoxy group. Para isomer is obtained in 90% yield.
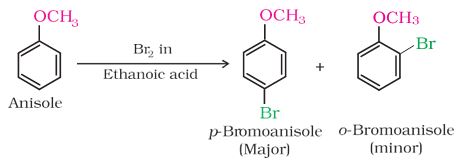
(2) Friedel-Crafts reaction: Anisole undergoes Friedel-Crafts reaction, i.e., the alkyl and acyl groups are introduced at ortho and para positions by reaction with alkyl halide and acyl halide in the presence of anhydrous aluminium chloride (a Lewis acid) as catalyst.
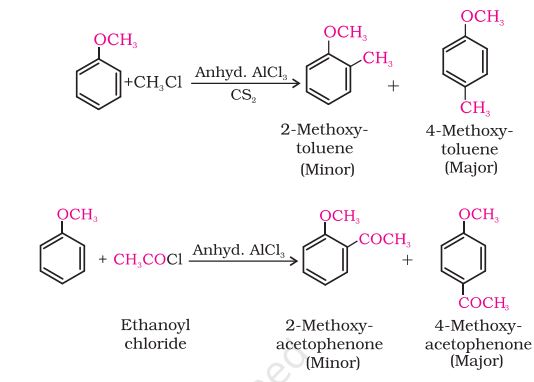
(3) Nitration: Anisole reacts with a mixture of concentrated sulphuric and nitric acids to yield a mixture of ortho and para nitroanisole.
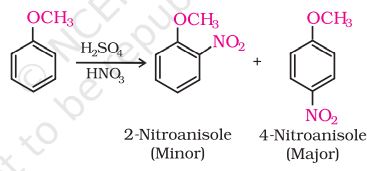
INTEXT QUESTION
Write the reactions of Williamson synthesis of 2-ethoxy-3-methylpentane starting from ethanol and 3-methylpentan-2-ol.
Which of the following is an appropriate set of reactants for the preparation of 1-methoxy-4-nitrobenzene and why?

Predict the products of the following reactions:
(i) CH3 - CH2 - CH2 - O – CH3 - HBr \(\to\)
-
Aldehydes, Ketones and Carboxylic Acids.12
In the previous Unit, you have studied organic compounds with functional groups containing carbon- oxygen single bond. In this Unit, we will study about the organic compounds containing carbon-oxygen double bond (>C=O) called carbonyl group, which is one of the most important functional groups in organic chemistry.
In aldehydes, the carbonyl group is bonded to a carbon and hydrogen while in the ketones, it is bonded to two carbon atoms. The carbonyl compounds in which carbon of carbonyl group is bonded to carbon or hydrogen and oxygen of hydroxyl moiety (-OH) are known as carboxylic acids, while in compounds where carbon is attached to carbon or hydrogen and nitrogen of -NH2 moiety or to halogens are called amides and acyl halides respectively. Esters and anhydrides are derivatives of carboxylic acids. The general formulas of these classes of compounds are given below:
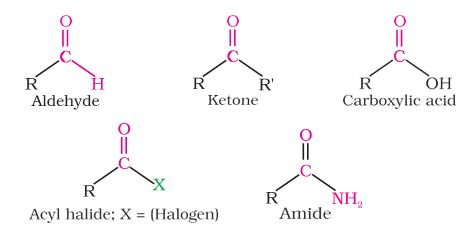
Aldehydes, ketones and carboxylic acids are widespread in plants and animal kingdom. They play an important role in biochemical processes of life. They add fragrance and flavour to nature, for example, vanillin (from vanilla beans), salicylaldehyde (from meadow sweet) and cinnamaldehyde (from cinnamon) have very pleasant fragrances.
They are used in many food products and pharmaceuticals to add flavours. Some of these families are manufactured for use as solvents (i.e., acetone) and for preparing materials like adhesives, paints, resins, perfumes, plastics, fabrics, etc.
Nomenclature and Structure of Carbonyl Group
Nomenclature
Aldehydes and ketones
Aldehydes and ketones are the simplest and most important carbonyl compounds.
There are two systems of nomenclature of aldehydes and ketones.
(a) Common names
Aldehydes and ketones are often called by their common names instead of IUPAC names. The common names of most aldehydes are derived from the common names of the corresponding carboxylic acids [Section 12.6.1] by replacing the ending –ic of acid with aldehyde. At the same time, the names reflect the Latin or Greek term for the original source of the acid or aldehyde. The location of the substituent in the carbon chain is indicated by Greek letters \(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGaeqySdeMaai % ilaiabek7aIjaacYcacqaHZoWzcaGGSaGaeqiTdqgaaa!3E92! \alpha ,\beta ,\gamma ,\delta \) etc. The \(\alpha\)-carbon being the one directly linked to the aldehyde group, \(\beta\)- carbon the next, and so on. For example
The common names of ketones are derived by naming two alkyl or aryl groups bonded to the carbonyl group. The locations of substituents are indicated by Greek letters, \(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGaeqySdeMafq % ySdeMbauaacaGGSaGaeqOSdiMafqOSdiMbauaaaaa!3D3E! \alpha \alpha ',\beta \beta '\) and so on beginning with the carbon atoms next to the carbonyl group, indicated as \(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGaeqySdeMafq % ySdeMbauaaaaa!3940! \alpha \alpha '\). Some ketones have historical common names, the simplest dimethyl ketone is called acetone. Alkyl phenyl ketones are usually named by adding the name of acyl group as prefix to the word phenone. For example
(b) IUPAC names
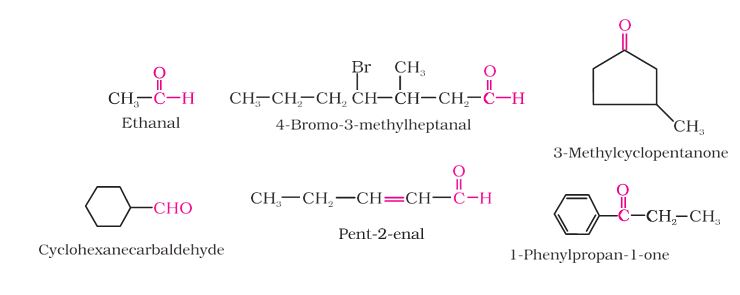
The common and IUPAC names of some aldehydes and ketones are given in Table 12.1.
Table 12.1: Common and IUPAC Names of Some Aldehydes and Ketones
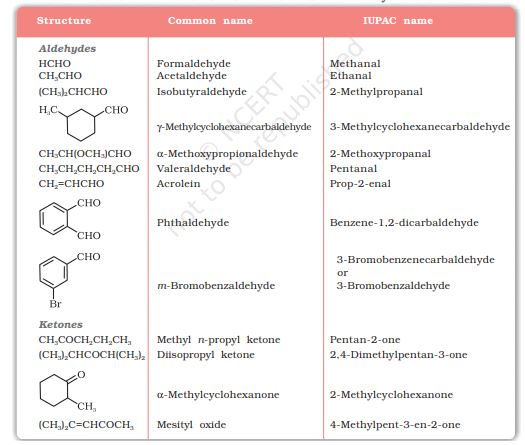
STRUCTURE of the Carbonyl Group
The carbonyl carbon atom is sp2-hybridised and forms three sigma (\(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGaeq4Wdmhaaa!37B9! \sigma \)) bonds. The fourth valence electron of carbon remains in its p-orbital and forms a \(\pi\)-bond with oxygen by overlap with p-orbital of an oxygen. In addition, the oxygen atom also has two non bonding electron pairs. Thus, the carbonyl carbon and the three atoms attached to it lie in the same plane and the \(\pi\)-electron cloud is above and below this plane. The bond angles are approximately 120° as expected of a trigonal coplanar structure (Figure 12.1).
Fig.12.1 Orbital diagram for the formation of carbonyl group
The carbon-oxygen double bond is polarised due to higher electronegativity of oxygen relative to carbon. Hence, the carbonyl carbon is an electrophilic (Lewis acid), and carbonyl oxygen, a nucleophilic (Lewis base) centre. Carbonyl compounds have substantial dipole moments and are polar than ethers. The high polarity of the carbonyl group is explained on the basis of resonance involving a neutral
(A) and a dipolar (B) structures as shown.
INTEXT SOLUTION
Write the structures of the following compounds.
\(\alpha\)-Methoxypropionaldehyde (ii) 3-Hydroxybutanal
(iii) 2-Hydroxycyclopentane carbaldehyde (iv) 4-Oxopentanal
(v) Di-sec. butyl ketone (vi) 4-Fluoroacetophenone
Preparation of Aldehydes and Ketones
Some important methods for the preparation of aldehydes and ketones are as follows:
Preparation of Aldehydes and Ketones
1. By oxidation of alcohols
Aldehydes and ketones are generally prepared by oxidation of primary and secondary alcohols, respectively (Unit 11, Class XII).
2. By dehydrogenation of alcohols
This method is suitable for volatile alcohols and is of industrial application. In this method alcohol vapours are passed over heavy metal catalysts (Ag or Cu). Primary and secondary alcohols give aldehydes and ketones, respectively (Unit 11, Class XII).
3. From hydrocarbons
(1) By ozonolysis of alkenes: As we know, ozonolysis of alkenes followed by reaction with zinc dust and water gives aldehydes,
ketones or a mixture of both depending on the substitution pattern of the alkene (Unit 13, Class XI).
(2) By hydration of alkynes: Addition of water to ethyne in the presence of H2SO4 and HgSO4 gives acetaldehyde. All other alkynes give ketones in this reaction (Unit 13, Class XI).
Preparation of Aldehydes
1. From acyl chloride (acid chloride)
Acyl chloride (acid chloride) is hydrogenated over catalyst, palladium on barium sulphate. This reaction is called Rosenmund reduction.
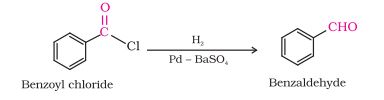
2. From nitriles and esters
Nitriles are reduced to corresponding imine with stannous chloride in the presence of hydrochloric acid, which on hydrolysis give corresponding aldehyde.
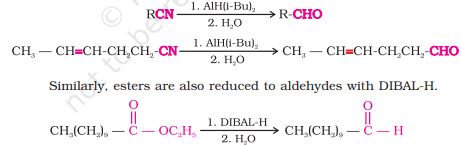
This reaction is called Stephen reaction.
Alternatively, nitriles are selectively reduced by diisobutylaluminium hydride, (DIBAL-H) to imines followed by hydrolysis to aldehydes:
3. From hydrocarbons
Aromatic aldehydes (benzaldehyde and its derivatives) are prepared from aromatic hydrocarbons by the following methods:
(1) By oxidation of methylbenzene
Strong oxidising agents oxidise toluene and its derivatives to benzoic acids. However, it is possible to stop the oxidation at the aldehyde stage with suitable reagents that convert the methyl group to an intermediate that is difficult to oxidise further. The following methods are used for this purpose.
(a) Use of chromyl chloride (CrO2Cl2): Chromyl chloride oxidises methyl group to a chromium complex, which on hydrolysis gives corresponding benzaldehyde.
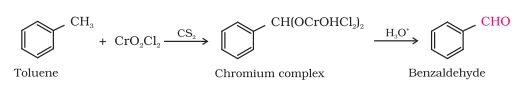
This reaction is called Etard reaction.
(b) Use of chromic oxide (CrO3): Toluene or substituted toluene is converted to benzylidene diacetate on treating with chromic oxide in acetic anhydride. The benzylidene diacetate can be hydrolysed to corresponding benzaldehyde with aqueous acid.
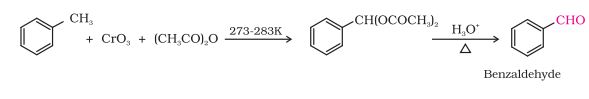
(2) By side chain chlorination followed by hydrolysis
Side chain chlorination of toluene gives benzal chloride, which on hydrolysis gives benzaldehyde. This is a commercial method of manufacture of benzaldehyde.
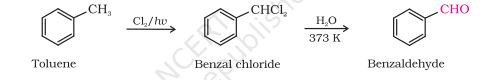
(3) By Gatterman – Koch reaction
When benzene or its derivative is treated with carbon monoxide and hydrogen chloride in the presence of anhydrous aluminium chloride or cuprous chloride, it gives benzaldehyde or substituted benzaldehyde.

1. From acyl chlorides
Treatment of acyl chlorides with dialkylcadmium, prepared by the reaction of cadmium chloride with Grignard reagent, gives ketones.
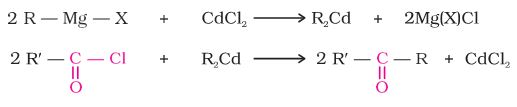
2. From nitriles
Treating a nitrile with Grignard reagent followed by hydrolysis yields a ketone.

3. From benzene or substituted benzenes
When benzene or substituted benzene is treated with acid chloride in the presence of anhydrous aluminium chloride, it affords the corresponding ketone. This reaction is known as Friedel-Crafts acylation reaction.
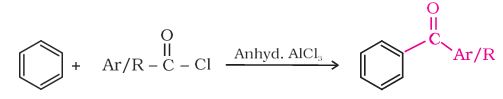
EXAMPLE 1
Give names of the reagents to bring about the following transformations:
(i) Hexan-1-ol to hexanal (ii) Cyclohexanol to cyclohexanone
(iii) p-Fluorotoluene to (iv) Ethanenitrile to ethanal
p-fluorobenzaldehyde
(v) Allyl alcohol to propenal (vi) But-2-ene to ethanal
SOLUTION
(i) C5H5NH+CrO3Cl-(PCC) (ii) Anhydrous CrO3
(iii) CrO3 in the presence of acetic anhydride/ (iv) (Diisobutyl)aluminium hydride (DIBAL-H)
1. CrO2Cl2 2. HOH
(v) PCC (vi) O3/H2O-Zn dust
INTEXT QUESTION
Write the structures of products of the following reactions;
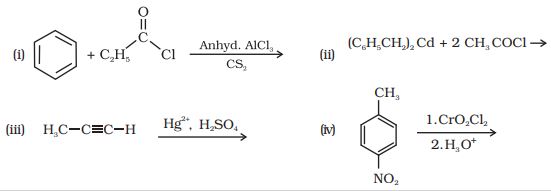
Physical Properties
The physical properties of aldehydes and ketones are described as follows.
Methanal is a gas at room temperature. Ethanal is a volatile liquid. Other aldehydes and ketones are liquid or solid at roomtemperature. The boiling points of aldehydes and ketones are higher than hydrocarbons and ethers of comparable molecular masses. It is due to weak molecular association in aldehydes and ketones arising out of the dipole-dipole interactions. Also, their boiling points are lower than those of alcohols of similar molecular masses due to absence of intermolecular hydrogen bonding. The following compounds of molecular masses 58 and 60 are ranked in order of increasing boiling points.
The lower members of aldehydes and ketones such as methanal, ethanal and propanone are miscible with water in all proportions, because they form hydrogen bond with water.

However, the solubility of aldehydes and ketones decreases rapidly on increasing the length of alkyl chain. All aldehydes and ketones are fairly soluble in organic solvents like benzene, ether, methanol, chloroform, etc. The lower aldehydes have sharp pungent odours. As the size of the molecule increases, the odour becomes less pungent and more fragrant. In fact, many naturally occurring aldehydes and ketones are used in the blending of perfumes and flavouring agents.
EXAMPLE 2
Arrange the following compounds in the increasing order of their boiling points:
CH3CH2CH2CHO, CH3CH2CH2CH2OH, H5C2-O-C2H5, CH3CH2CH2CH3
SOLUTION
The molecular masses of these compounds are in the range of 72 to 74. Since only butan-1-ol molecules are associated due to extensive intermolecular hydrogen bonding, therefore, the boiling point of butan-1-ol would be the highest. Butanal is more polar than ethoxyethane. Therefore, the intermolecular dipole-dipole attraction is stronger in the former. n-Pentane molecules have only weak van der Waals forces. Hence increasing order of boiling points of the given compounds is as follows:
CH3CH2CH2CH3 < H5C2-O-C2H5 < CH3CH2CH2CHO < CH3CH2CH2CH2OH
INTEXT QUESTION
Arrange the following compounds in increasing order of their boiling points.
CH3CHO, CH3CH2OH, CH3OCH3, CH3CH2CH3
Chemical Reactions
Since aldehydes and ketones both possess the carbonyl functional group, they undergo similar chemical reactions.
1. Nucleophilic addition reactions
Contrary to electrophilic addition reactions observed in alkenes (refer Unit 13, Class XI), the aldehydes and ketones undergo nucleophilic addition reactions.
(1) Mechanism of nucleophilic addition reactions
A nucleophile attacks the electrophilic carbon atom of the polar carbonyl group from a direction approximately perpendicular to the plane of sp2 hybridised orbitals of carbonyl carbon (Fig. 12.2).
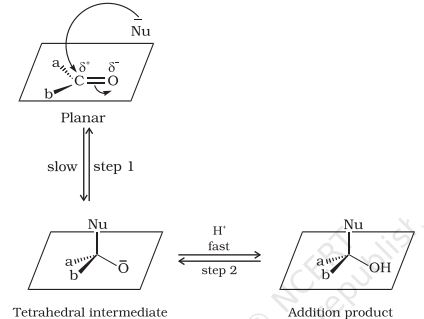
Fig.12.2: Nucleophilic attack on carbonyl carbon
The hybridisation of carbon changes from sp2 to sp3 in this process, and a tetrahedral alkoxide intermediate is produced. This intermediate captures a proton from the reaction medium to give the electrically neutral product. The net result is addition of Nu– and H+ across the carbon oxygen double bond as shown in Fig. 12.2.
(2) Reactivity
Aldehydes are generally more reactive than ketones in nucleophilic addition reactions due to steric and electronic reasons. Sterically, the presence of two relatively large substituents in ketones hinders the approach of nucleophile to carbonyl carbon than in aldehydes having only one such substituent. Electronically, aldehydes are more reactive than ketones because two alkyl groups reduce the electrophilicity of the carbonyl carbon more effectively than in former.
EXAMPLE 3
Would you expect benzaldehyde to be more reactive or less reactive in nucleophilic addition reactions than propanal? Explain your answer.
SOLUTION
The carbon atom of the carbonyl group of benzaldehyde is less electrophilic than carbon atom of the carbonyl group present in
propanal. The polarity of the carbonyl group is reduced in benzaldehyde due to resonance as shown below and hence it is less reactive than propanal.
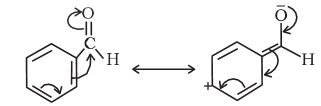
(1) Some important examples of nucleophilic addition and nucleophilic addition-elimination reactions:
(a) Addition of hydrogen cyanide (HCN):
Aldehydes and ketones react with hydrogen cyanide (HCN) to yield cyanohydrins. This reaction occurs very slowly with pure HCN. Therefore, it is catalysed by a base and the generated cyanide ion (CN-) being a stronger nucleophile readily adds to carbonyl compounds to yield corresponding cyanohydrin. Cyanohydrins are useful synthetic intermediates.
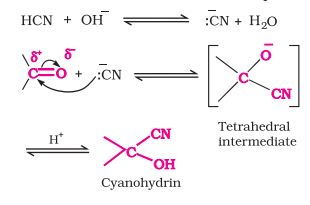
(b) Addition of sodium hydrogensulphite: Sodium hydrogensulphite adds to aldehydes and ketones to form the addition products.
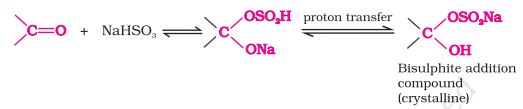
The position of the equilibrium lies largely to the right hand side for most aldehydes and to the left for mostketones due to steric reasons. The hydrogensulphite addition compound is water soluble and can be converted back to the original carbonyl compound by treating it with dilute mineral acid or alkali. Therefore, these are useful for separation and purification of aldehydes.
(c) Addition of Grignard reagents: (refer Unit 11, Class XII).
(d) Addition of alcohols: Aldehydes react with one equivalent of monohydric alcohol in the presence of dry hydrogen chloride to yield alkoxyalcohol intermediate, known as hemiacetals, which further react with one more molecule of alcohol to give a gem-dialkoxy compound known as acetal as shown in the reaction.
Ketones react with ethylene glycol under similar conditions to form cyclic products known as ethylene glycol ketals.
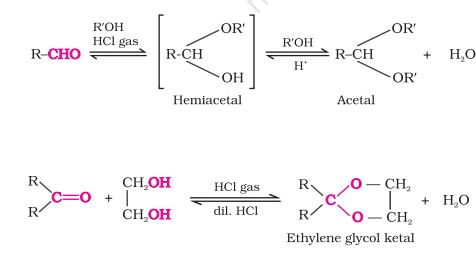
Dry hydrogen chloride protonates the oxygen of the carbonyl compounds and therefore, increases the electrophilicity of the carbonyl carbon facilitating the nucleophilic attack of ethylene glycol. Acetals and ketals are hydrolysed with aqueous mineral acids to yield corresponding aldehydes and ketones respectively.
(e) Addition of ammonia and its derivatives: Nucleophiles, such as ammonia and its derivatives H2N-Z add to the carbonyl group of aldehydes and ketones. The reaction is reversible and catalysed by acid. The equilibrium favours the product formation due to rapid dehydration of the intermediate to form >C=N-Z.
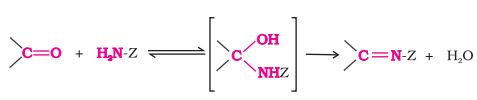
Z = Alkyl, aryl, OH, NH2, C6H5NH, NHCONH2, etc.
Table 12.2: Some N-Substituted Derivatives of Aldehydes and Ketones (>C=N-Z)
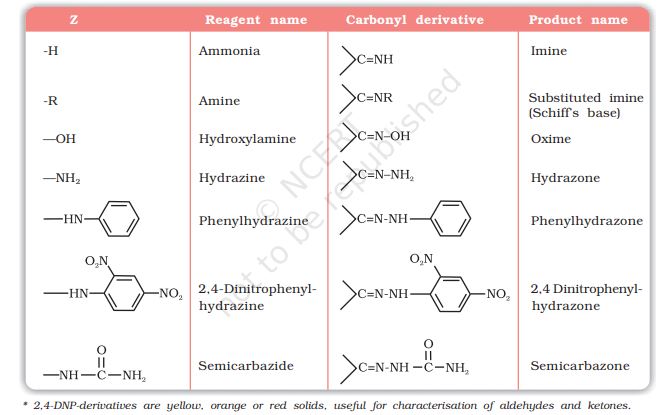
2. Reduction
(1) Reduction to alcohols: Aldehydes and ketones are reduced to primary and secondary alcohols respectively by sodium borohydride (NaBH4) or lithium aluminium hydride (LiAlH4) as well as by catalytic hydrogenation (Unit 11, Class XII).
(2) Reduction to hydrocarbons: The carbonyl group of aldehydes and ketones is reduced to CH2 group on treatment with zinc- amalgam and concentrated hydrochloric acid [Clemmensen reduction] or with hydrazine followed by heating with sodium or potassium hydroxide in high boiling solvent such as ethylene glycol (Wolff-Kishner reduction).
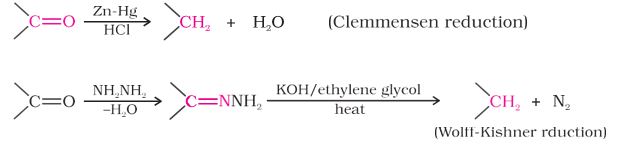
3. Oxidation
Aldehydes differ from ketones in their oxidation reactions. Aldehydes are easily oxidised to carboxylic acids on treatment with common oxidising agents like nitric acid, potassium permanganate, potassium dichromate, etc. Even mild oxidising agents, mainly Tollens’ reagent and Fehlings’ reagent also oxidise aldehydes.
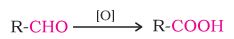
Ketones are generally oxidised under vigorous conditions, i.e., strong oxidising agents and at elevated temperatures. Their oxidation involves carbon-carbon bond cleavage to afford a mixture of carboxylic acids having lesser number of carbon atoms than the parent ketone.

The mild oxidising agents given below are used to distinguish aldehydes from ketones:
(1) Tollens’ test: On warming an aldehyde with freshly prepared ammoniacal silver nitrate solution (Tollens’ reagent), a bright silver mirror is produced due to the formation of silver metal. The aldehydes are oxidised to corresponding carboxylate anion. The reaction occurs in alkaline medium.

(2) Fehling’s test: Fehling reagent comprises of two solutions, Fehling solution A and Fehling solution B. Fehling solution A is aqueous copper sulphate and Fehling solution B is alkaline sodium potassium tartarate (Rochelle salt). These two solutions are mixed in equal amounts before test. On heating an aldehyde with Fehling’s reagent, a reddish brown precipitate is obtained. Aldehydes are oxidised to corresponding carboxylate anion. Aromatic aldehydes do not respond to this test.
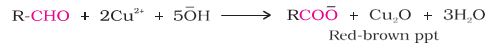
(3) Oxidation of methyl ketones by haloform reaction: Aldehydes and ketones having at least one methyl group linked to the carbonyl carbon atom (methyl ketones) are oxidised by sodium hypohalite to sodium salts of corresponding carboxylic acids having one carbon atom less than that of carbonyl compound. The methyl group is converted to haloform. This oxidation does not affect a carbon-carbon double bond, if present in the molecule.
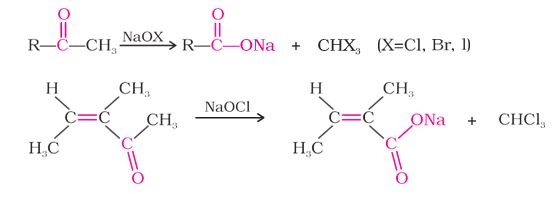
Iodoform reaction with sodium hypoiodite is also used for detection of CH3CO group or CH3CH(OH) group which produces CH3CO group on oxidation.
EXAMPLE 4
An organic compound (A) with molecular formula C8H8O forms an orange-red precipitate with 2,4-DNP reagent and gives yellow precipitate on heating with iodine in the presence of sodium hydroxide. It neither reduces Tollens’ or Fehlings’ reagent, nor does it decolourise bromine water or Baeyer’s reagent. On drastic oxidation with chromic acid, it gives a carboxylic acid (B) having molecular formula C7H6O2. Identify the compounds (A) and (B) and explain thereactions involved.
SOLUTION
(A) forms 2,4-DNP derivative. Therefore, it is an aldehyde or a ketone. Since it does not reduce Tollens’ or Fehling reagent, (A) must be a ketone.
(A) responds to iodoform test. Therefore, it should be a methyl ketone. The molecular formula of (A) indicates high degree of unsaturation, yet it does not decolourise bromine water or Baeyer’s reagent. This indicates the presence of unsaturation due to an aromatic ring.
Compound (B), being an oxidation product of a ketone should be a carboxylic acid. The molecular formula of (B) indicates that it should be benzoic acid and compound (A) should, therefore, be a monosubstituted aromatic methyl ketone. The molecular formula of
(A) indicates that it should be phenyl methyl ketone (acetophenone). Reactions are as follows:
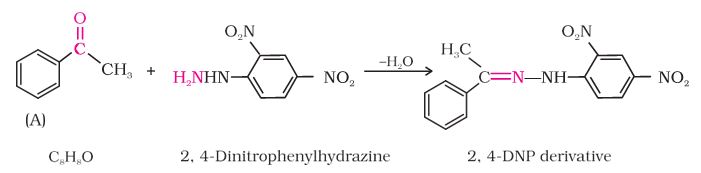
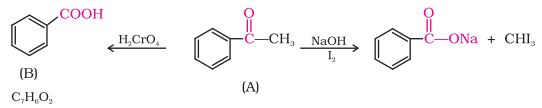
4. Reactions due to a-hydrogen
Acidity of \(\alpha\)-hydrogens of aldehydes and ketones: The aldehydes and ketones undergo a number of reactions due to the acidic nature of -hydrogen.
The acidity of \(\alpha\)-hydrogen atoms of carbonyl compounds is due to the strong electron withdrawing effect of the carbonyl group and resonance stabilisation of the conjugate base.
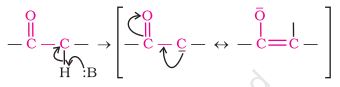
(1) Aldol condensation: Aldehydes and ketones having at least one \(\alpha\)-hydrogen undergo a reaction in the presence of dilute alkali as catalyst to form \(\beta\)-hydroxy aldehydes (aldol) or \(\beta\)-hydroxy ketones (ketol), respectively. This is known as Aldol reaction.
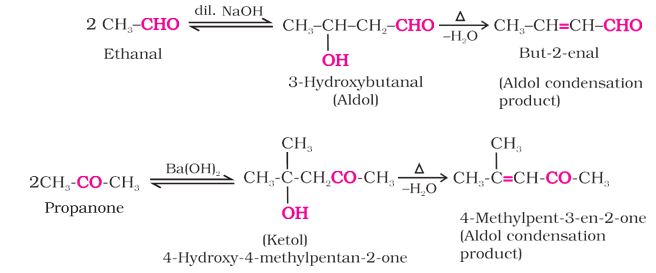
The name aldol is derived from the names of the two functional groups, aldehyde and alcohol, present in the products. The aldol and ketol readily lose water to give \(\alpha\),\(\beta\)-unsaturated carbonyl compounds which are aldol condensation products and the reaction is called Aldol condensation. Though ketones give ketols (compounds containing a keto and alcohol groups), the general name aldol condensation still applies to the reactions of ketones due to their similarity with aldehydes.
(2) Cross aldol condensation: When aldol condensation is carried out between two different aldehydes and / or ketones, it is called cross aldol condensation. If both of them contain \(\alpha\)-hydrogen atoms, it gives a mixture of four products. This is illustrated below by aldol reaction of a mixture of ethanal and propanal.
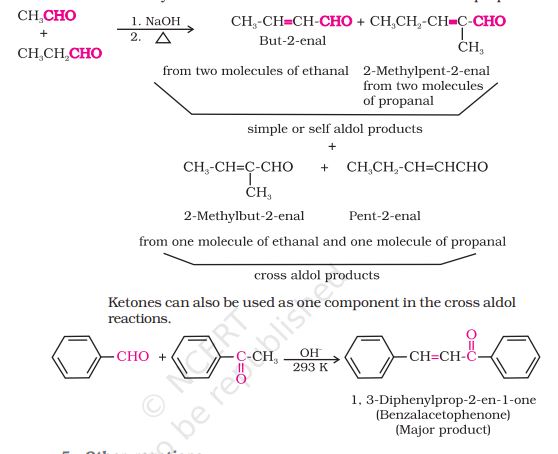
Ketones can also be used as one component in the cross aldol reactions.
5. Other reactions
(1) Cannizzaro reaction: Aldehydes which do not have an \(\alpha\)-hydrogen atom, undergo self oxidation and reduction (disproportionation) reaction on heating with concentrated alkali. In this reaction, one molecule of the aldehyde is reduced to alcohol while another is oxidised to carboxylic acid salt.
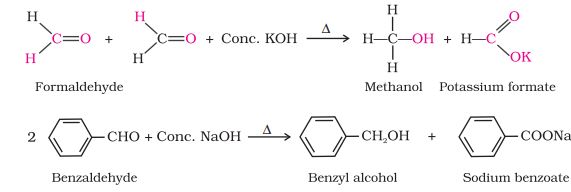
(2) Electrophilic substitution reaction: Aromatic aldehydes and ketones undergo electrophilic substitution at the ring in which the carbonyl group acts as a deactivating and meta-directing group.
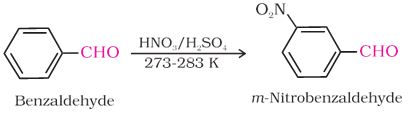
INTEXT QUESTION
Arrange the following compounds in increasing order of their reactivity in nucleophilic addition reactions.
(1) Ethanal, Propanal, Propanone, Butanone.
(2)Benzaldehyde, p-Tolualdehyde, p-Nitrobenzaldehyde, Acetophenone.
Hint: Consider steric effect and electronic effect.
Predict the products of the following reactions:
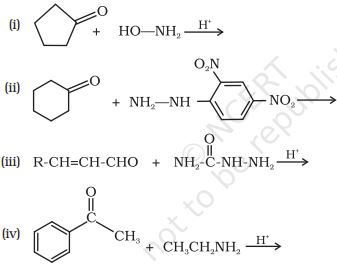
Uses of Aldehydes and Ketones
In chemical industry aldehydes and ketones are used as solvents, starting materials and reagents for the synthesis of other products. Formaldehyde is well known as formalin (40%) solution used to preserve biological specimens and to prepare bakelite (a phenol-formaldehyde resin), urea-formaldehyde glues and other polymeric products. Acetaldehyde is used primarily as a starting material in the manufacture of acetic acid, ethyl acetate, vinyl acetate, polymers and drugs. Benzaldehyde is used in perfumery and in dye industries. Acetone and ethyl methyl ketone are common industrial solvents. Many aldehydes and ketones, e.g., butyraldehyde, vanillin, acetophenone, camphor, etc. are well known for their odours and flavours.
Carboxylic Acids
Carbon compounds containing a carboxyl functional group, –COOH are called carboxylic acids. The carboxyl group, consists of a carbonyl group attached to a hydroxyl group, hence its name carboxyl. Carboxylic acids may be aliphatic (RCOOH) or aromatic (ArCOOH) depending on the group, alkyl or aryl, attached to carboxylic carbon. Large number of carboxylic acids are found in nature. Some higher members of aliphatic carboxylic acids (C12 – C18) known as fatty acids, occur in natural fats as esters of glycerol. Carboxylic acids serve as starting material for several other important organic compounds such as anhydrides, esters, acid chlorides, amides, etc.
Nomenclature and Structure of Carboxyl Group
Nomenclature
Since carboxylic acids are amongst the earliest organic compounds to be isolated from nature, a large number of them are known by their common names. The common names end with the suffix –ic acid and have been derived from Latin or Greek names of their natural sources. For example, formic acid (HCOOH) was first obtained from red ants (Latin: formica means ant), acetic acid (CH3COOH) from vinegar (Latin: acetum, means vinegar), butyric acid (CH3CH2CH2COOH) from rancid butter (Latin: butyrum, means butter).
In the IUPAC system, aliphatic carboxylic acids are named by replacing the ending –e in the name of the corresponding alkane with – oic acid. In numbering the carbon chain, the carboxylic carbon is numbered one. For naming compounds containing more than one carboxyl group, the alkyl chain leaving carboxyl groups is numbered and the number of carboxyl groups is indicated by adding the multiplicative prefix, dicarboxylic acid, tricarboxylic acid, etc. to the name of parent alkyl chain. The position of –COOH groups are indicated by the arabic numeral before the multiplicative prefix. Some of the carboxylic acids along with their common and IUPAC names are listed in Table 12.3.
Table 12.3 Names and Structures of Some Carboxylic Acids
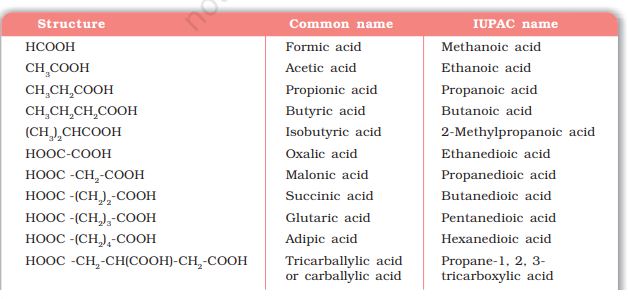

Structure of Carboxyl Group
In carboxylic acids, the bonds to the carboxyl carbon lie in one plane and are separated by about 120°. The carboxylic carbon is less electrophilic than carbonyl carbon because of the possible resonance structure shown below:
INTEXT QUESTION
Give the IUPAC names of the following compounds:
(i) Ph CH2CH2COOH (ii) (CH3)2C=CHCOOH
Methods OF Preparation of Carboxylic Acids
Some important methods of preparation of carboxylic acids are as follows.
1. From primary alcohols and aldehydes
Primary alcohols are readily oxidised to carboxylic acids with common oxidising agents such as potassium permanganate (KMnO4) in neutral, acidic or alkaline media or by potassium dichromate (K2Cr2O7) and chromium trioxide (CrO3) in acidic media (Jones reagent).
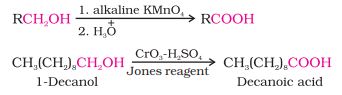
Carboxylic acids are also prepared from aldehydes by the use of mild oxidising agents (Section 12.4).
2. From alkylbenzenes
Aromatic carboxylic acids can be prepared by vigorous oxidation of alkyl benzenes with chromic acid or acidic or alkaline potassium permanganate. The entire side chain is oxidised to the carboxyl group irrespective of length of the side chain. Primary and secondary alkyl groups are oxidised in this manner while tertiary group is not affected. Suitably substituted alkenes are also oxidised to carboxylic acids with these oxidising reagents (refer Unit 13, Class XI).
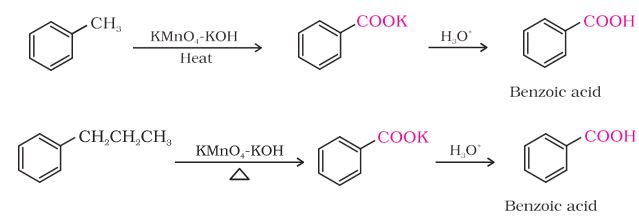
3.From nitriles and amides
Nitriles are hydrolysed to amides and then to acids in the presence of H+ or \(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaWaa0aaaeaaca % WGpbaaaaaa!36DB! \overline O \)H as catalyst. Mild reaction conditions are used to stop thereaction at the amide stage.
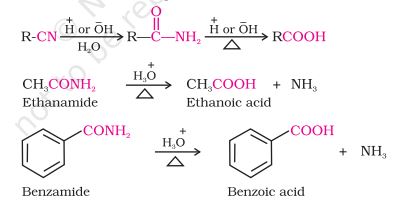
4. From Grignard reagents
Grignard reagents react with carbon dioxide (dry ice) to form salts of carboxylic acids which in turn give corresponding carboxylic acids after acidification with mineral acid.
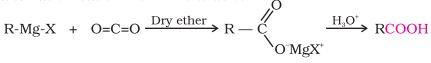
As we know, the Grignard reagents and nitriles can be prepared from alkyl halides (refer Unit 10, Class XII). The above methods
(3 and 4) are useful for converting alkyl halides into corresponding carboxylic acids having one carbon atom more than that present in alkyl halides (ascending the series).5. From acyl halides and anhydrides
Acid chlorides when hydrolysed with water give carboxylic acids or more readily hydrolysed with aqueous base to give carboxylate ions which on acidification provide corresponding carboxylic acids. Anhydrides on the other hand are hydrolysed to corresponding acid(s) with water.
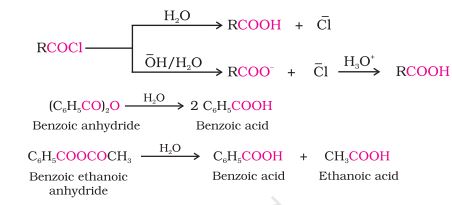
6. From esters
Acidic hydrolysis of esters gives directly carboxylic acids while basic hydrolysis gives carboxylates, which on acidification give corresponding carboxylic acids.
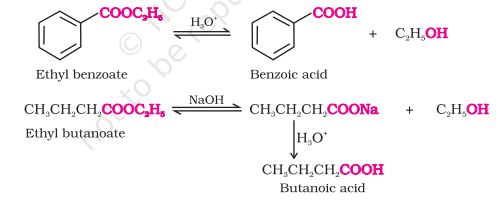
EXAMPLE 5
Write chemical reactions to affect the following transformations:
Butan-1-ol to butanoic acid
Benzyl alcohol to phenylethanoic acid
3-Nitrobromobenzene to 3-nitrobenzoic acid
4-Methylacetophenone to benzene-1,4-dicarboxylic acid
Cyclohexene to hexane-1,6-dioic acid
Butanal to butanoic acid.
SOLUTION
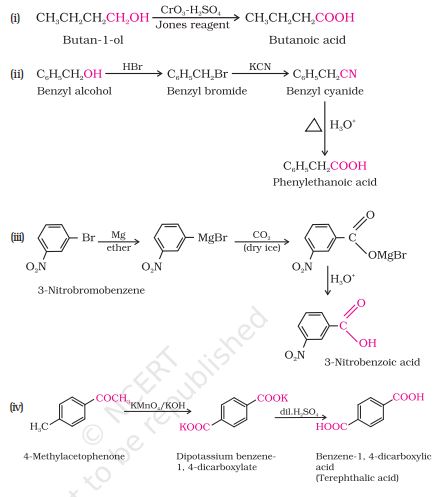
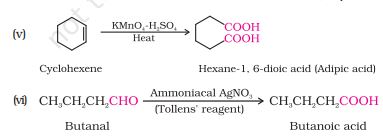
INTEXT QUESTION
Show how each of the following compounds can be converted to benzoic acid.
(i) Ethylbenzene (ii) Acetophenone
(iii) Bromobenzene (iv) Phenylethene (Styrene)
Physical Properties
Aliphatic carboxylic acids upto nine carbon atoms are colourless liquids at room temperature with unpleasant odours. The higher acids are wax like solids and are practically odourless due to their low volatility. Carboxylic acids are higher boiling liquids than aldehydes, ketones and even alcohols of comparable molecular masses. This is due to more extensive association of carboxylic acid molecules through intermolecular hydrogen bonding. The hydrogen bonds are not broken completely even in the vapour phase. In fact,most carboxylic acids exist as dimer in the vapour phase or in the aprotic solvents.
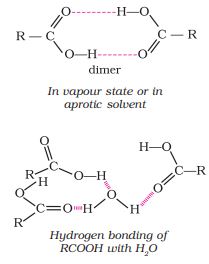
Simple aliphatic carboxylic acids having upto four carbon atoms are miscible in water due to the formation of hydrogen bonds with water. The solubility decreases with increasing number of carbon atoms. Higher carboxylic acids are practically insoluble in water due to the increased hydrophobic interaction of hydrocarbon part. Benzoic acid, the simplest aromatic carboxylic acid is nearly insoluble in cold water. Carboxylic acids are also soluble in less polar organic solvents like benzene, ether, alcohol, chloroform, etc.
Chemical Reactions
The reaction of carboxylic acids are classified as follows:
Acidity
Reactions with metals and alkalies
The carboxylic acids like alcohols evolve hydrogen with electropositive metals and form salts with alkalies similar to phenols. However, unlike phenols they react with weaker bases such as carbonates and hydrogencarbonates to evolve carbon dioxide. This reaction is used to detect the presence of carboxyl group in an organic compound.
Involving Cleavage of O–H Bond

Carboxylic acids dissociate in water to give resonance stabilised carboxylate anions and hydronium ion.
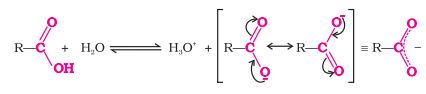
For the above reaction:
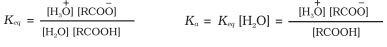
where Keq, is equilibrium constant and Ka is the acid dissociation constant.
For convenience, the strength of an acid is generally indicated by its pKa value rather than its Ka value.
pKa = – log Ka
The pKa of hydrochloric acid is –7.0, where as pKa of trifluoroacetic acid (the strongest carboxylic acid), benzoic acid and acetic acid are 0.23, 4.19 and 4.76, respectively.
Smaller the pKa, the stronger the acid ( the better it is as a proton donor). Strong acids have pKa values < 1, the acids with pKa values between 1 and 5 are considered to be moderately strong acids, weak acids have pKa values between 5 and 15, and extremely weak acids have pKa values >15.
Carboxylic acids are weaker than mineral acids, but they are stronger acids than alcohols and many simple phenols (pKa is ~16 for ethanol and 10 for phenol). In fact, carboxylic acids are amongst the most acidic organic compounds you have studied so far. You already know why phenols are more acidic than alcohols. The higher acidity of carboxylic acids as compared to phenols can be understood similarly. The conjugate base of carboxylic acid, a carboxylate ion, is stabilised by two equivalent resonance structures in which the negative charge is at the more electronegative oxygen atom. The conjugate base of phenol, a phenoxide ion, has non-equivalent resonance structures in which the negative charge is at the less electronegative carbon atom. Therefore, resonance in phenoxide ion is not as important as it is in carboxylate ion. Further, the negative charge is delocalised over two electronegative oxygen atoms in carboxylate ion whereas it is less effectively delocalised over one oxygen atom and less electronegative carbon atoms in phenoxide ion (Unit 11, Class XII). Thus, the carboxylate ion is more stabilised than phenoxide ion, so carboxylic acids are more acidic than phenols.
Effect of substituents on the acidity of carboxylic acids: Substituents may affect the stability of the conjugate base and thus, also affect the acidity of the carboxylic acids. Electron withdrawing groups increase the acidity of carboxylic acids by stabilising the conjugate base through delocalisation of the negative charge by inductive and/or resonance effects. Conversely, electron donating groups decrease the acidity by destabilising the conjugate base.
Electron withdrawing group (EWG) stabilises the carboxylate anion and strengthens the acid
Electron donating group (EDG) destabilises the carboxylate anion and weakens the acid
The effect of the following groups in increasing acidity order isPh < I < Br < Cl < F < CN < NO2 < CF3
Thus, the following acids are arranged in order of increasing acidity (based on pKa values):
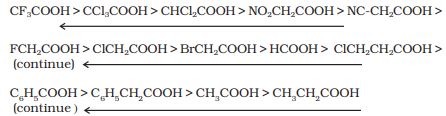
Direct attachment of groups such as phenyl or vinyl to the carboxylic acid, increases the acidity of corresponding carboxylic acid, contrary to the decrease expected due to resonance effect shown below:

This is because of greater electronegativity of sp2 hybridised carbon to which carboxyl carbon is attached. The presence of electron withdrawing group on the phenyl of aromatic carboxylic acid increases their acidity while electron donating groups decrease their acidity.

Reaction Involving Cleavage of C–OH Bond
1. Formation of anhydride
Carboxylic acids on heating with mineral acids such as H2SO4 or with P2O5 give corresponding anhydride.
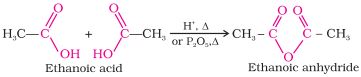
2.Esterification
Carboxylic acids are esterified with alcohols or phenols in the presence of a mineral acid such as concentrated H2SO4 or HCl gas as a catalyst.

Mechanism of esterification of carboxylic acids: The esterification of carboxylicacids with alcohols is a kind of nucleophilic acyl substitution. Protonation of the carbonyl oxygen activates the carbonyl group towards nucleophilic addition of the alcohol. Proton transfer in the tetrahedral intermediate converts the hydroxyl group into – +OH2 group, which, being a better leaving group, is eliminated as neutral water molecule. The protonated ester so formed finally loses a proton to give the ester.
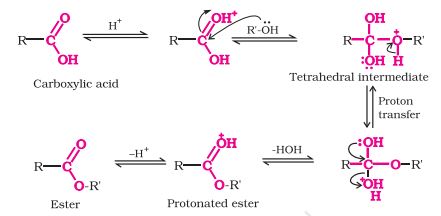
3. Reactions with PCl5, PCl3 and SOCl2
The hydroxyl group of carboxylic acids, behaves like that of alcohols and is easily replaced by chlorine atom on treating with PCl5, PCl3 or SOCl2. Thionyl chloride (SOCl2) is preferred because the other two products are gaseous and escape the reaction mixture making the purification of the products easier.
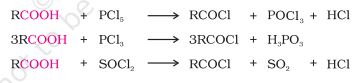
4. Reaction with ammonia
Carboxylic acids react with ammonia to give ammonium salt which on further heating at high temperature give amides. For example:

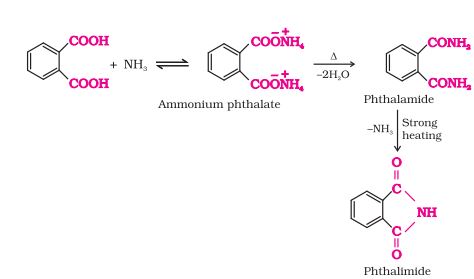
Reaction Involving -COOH Group
1. Reduction
Carboxylic acids are reduced to primary alcohols by lithium aluminium hydride or better with diborane. Diborane does not easily reduce functional groups such as ester, nitro, halo, etc. Sodium borohydride does not reduce the carboxyl group.

2. Decarboxylation
Carboxylic acids lose carbon dioxide to form hydrocarbons when their sodium salts are heated with sodalime (NaOH and CaO in the ratio of 3 : 1). The reaction is known as decarboxylation.

Alkali metal salts of carboxylic acids also undergo decarboxylation on electrolysis of their aqueous solutions and form hydrocarbons having twice the number of carbon atoms present in the alkyl group of the acid. The reaction is known as Kolbe electrolysis (Unit 13, Class XI).4.
Substitution Reactions in the Hydrocarbon Part
1. Halogenation
Carboxylic acids having an \(\alpha\)-hydrogen are halogenated at the -position on treatment with chlorine or bromine in the presence of small amount of red phosphorus to give \(\alpha\)-halocarboxylic acids. The reaction is known as Hell-Volhard-Zelinsky reaction.
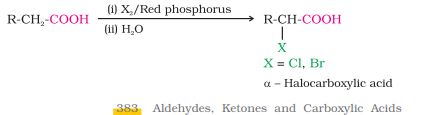
2. Ring substitution
Aromatic carboxylic acids undergo electrophilic substitution reactions in which the carboxyl group acts as a deactivating and meta-directing group. They however, do not undergo Friedel-Crafts reaction (because the carboxyl group is deactivating and the catalyst aluminium chloride (Lewis acid) gets bonded to the carboxyl group).
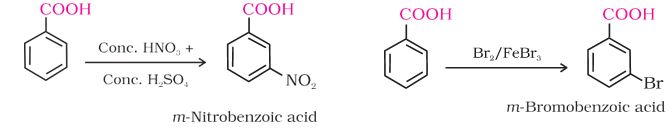
INTEXT QUESTION
Which acid of each pair shown here would you expect to be stronger?
(i) CH3CO2H or CH2FCO2H (ii) CH2FCO2H or CH2ClCO2H
(iii) CH2FCH2CH2CO2H or CH3CHFCH2CO2H
(4)

Uses OF Carboxylic Acids
Methanoic acid is used in rubber, textile, dyeing, leather and electroplating industries. Ethanoic acid is used as solvent and as vinegar in food industry. Hexanedioic acid is used in the manufacture of nylon-6, 6. Esters of benzoic acid are used in perfumery. Sodium benzoate is used as a food preservative. Higher fatty acids are used for the manufacture of soaps and detergents.
-
Amines.13
Amines constitute an important class of organic compounds derived by replacing one or more hydrogen atoms of ammonia molecule by alkyl/aryl group(s). In nature, they occur among proteins, vitamins, alkaloids and hormones. Synthetic examples include polymers, dye stuffs and drugs. Two biologically active compounds, namely adrenaline and ephedrine, both containing secondary amino group, are used to increase blood pressure. Novocain, a synthetic amino compound, is used as an anaesthetic in dentistry. Benadryl, a well known antihistaminic drug also contains tertiary amino group. Quaternary ammonium salts are used as surfactants. Diazonium salts are intermediates in the preparation of a variety of aromatic compounds including dyes. In this Unit, you will learn about amines and diazonium salts.
1.AMINES
Amines can be considered as derivatives of ammonia, obtained by replacement of one, two or all the three hydrogen atoms by alkyl and/or aryl groups.
For example:
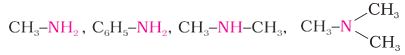
Structure of Amines
Like ammonia, nitrogen atom of amines is trivalent and carries an unshared pair of electrons. Nitrogen orbitals in amines are therefore, sp3 hybridised and the geometry of amines is pyramidal. Each of the three sp3 hybridised orbitals of nitrogen overlap with orbitals of hydrogen or carbon depending upon the composition of the amines. The fourth orbital of nitrogen in all amines contains an unshared pair of electrons. Due to the presence of unshared pair of electrons, the angle C–N–E, (where E is C or H) is less than 109.5°; for instance, it is 108o in case of trimethylamine as shown in Fig. 13.1.
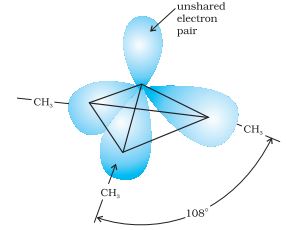
Fig. 13.1 Pyramidal shape of trimethylamine
Classification
Amines are classified as primary (1o), secondary (2o) and tertiary (3o) depending upon the number of hydrogen atoms replaced by alkyl or aryl groups in ammonia molecule. If one hydrogen atom of ammonia is replaced by R or Ar , we get RNH2 or ArNH , a primary amine (1o). If two hydrogen atoms of ammonia or one hydrogen atom of R-NH2 are replaced by another alkyl/aryl(R’) group, what would you get? Youget R-NHR’, secondary amine. The second alkyl/aryl group may be same or different. Replacement of another hydrogen atom by alkyl/aryl group leads to the formation of tertiary amine. Amines are said to be ‘simple’ when all the alkyl or aryl groups are the same, and ‘mixed’ when they are different.
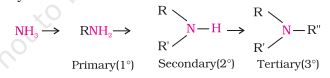
Nomenclature
In common system, an aliphatic amine is named by prefixing alkyl group to amine, i.e., alkylamine as one word (e.g., methylamine). In secondary and tertiary amines, when two or more groups are the same, the prefix di or tri is appended before the name of alkyl group. In IUPAC system, primary amines are named as alkanamines. The name is derived by replacement of ‘e’ of alkane by the word amine. For example, CH3NH2 is named as methanamine. In case, more than one amino group is present at different positions in the parent chain, their positions are specified by giving numbers to the carbon atoms bearing –NH2 groups and suitable prefix such as di, tri, etc. is attached to the amine. The letter ‘e’ of the suffix of the hydrocarbon part is retained. For example, H2N–CH2–CH2–NH2 is named as ethane-1, 2-diamine.
To name secondary and tertiary amines, we use locant N to designate substituent attached to a nitrogen atom. For example, CH3NHCH2CH3 is
named as N-methylethanamine and (CH3CH2)3N is named as N, N- diethylethanamine. More examples are given in Table 13.1.Table 13.1: Nomenclature of Some Alkylamines and Arylamines

In arylamines, –NH2 group is directly attached to the benzene ring. C6H5NH2 is the simplest example of arylamine. In common system, it is known as aniline. It is also an accepted IUPAC name. While naming arylamines according to IUPAC system, suffix ‘e’ of arene is replaced by ‘amine’. Thus in IUPAC system, C6H5–NH2 is named as benzenamine. Common and IUPAC names of some alkylamines and arylamines are given in Table 13.1.
INTEXT QUESTION
Classify the following amines as primary, secondary or tertiary:
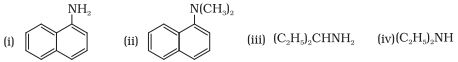
1. Write structures of different isomeric amines corresponding to the molecular formula, C4H11N.
2. Write IUPAC names of all the isomers.
3. What type of isomerism is exhibited by different pairs of amines?
Preparation of Amines
Amines are prepared by the following methods:
1. Reduction of nitro compounds
Nitro compounds are reduced to amines by passing hydrogen gas in the presence of finely divided nickel, palladium or platinum and also by reduction with metals in acidic medium. Nitroalkanes can also be similarly reduced to the corresponding alkanamines.
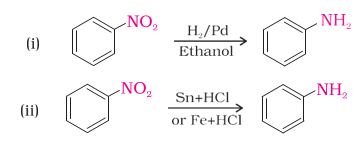
Reduction with iron scrap and hydrochloric acid is preferred because FeCl2 formed gets hydrolysed to release hydrochloric acid during the reaction. Thus, only a small amount of hydrochloric acid is required to initiate the reaction.
2. Ammonolysis of alkyl halides
You have read (Unit 10, Class XII) that the carbon - halogen bond in alkyl or benzyl halides can be easily cleaved by a nucleophile. Hence, an alkyl or benzyl halide on reaction with an ethanolic solution of ammonia undergoes nucleophilic substitution reaction in which the halogen atom is replaced by an amino (–NH2) group. This process of cleavage of the C–X bond by ammonia molecule is known as ammonolysis. The reaction is carried out in a sealed tube at 373 K. The primary amine thus obtained behaves as a nucleophile and can further react with alkyl halide to form secondary and tertiary amines, and finally quaternary ammonium salt.
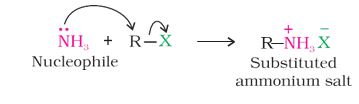
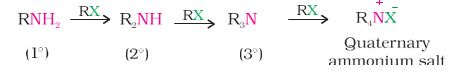
The free amine can be obtained from the ammonium salt by treatment with a strong base:
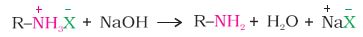
Ammonolysis has the disadvantage of yielding a mixture of primary, secondary and tertiary amines and also a quaternary ammonium salt. However, primary amine is obtained as a major product by taking large excess of ammonia.
The order of reactivity of halides with amines is RI > RBr >RCl.
EXAMPLE 1
Write chemical equations for the following reactions:
1. Reaction of ethanolic NH3 with C2H5Cl.
2. Ammonolysis of benzyl chloride and reaction of amine so formed with two moles of CH3Cl.
SOLUTION
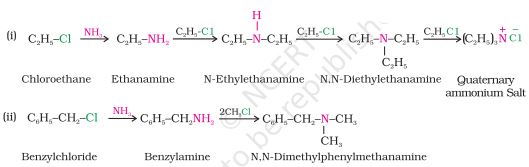
3. Reduction of nitriles
Nitriles on reduction with lithium aluminium hydride (LiAlH4) or catalytic hydrogenation produce primary amines. This reaction is used for ascent of amine series, i.e., for preparation of amines containing one carbon atom more than the starting amine.

4. Reduction of amides
The amides on reduction with lithium aluminium hydride yield amines.
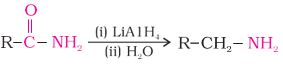
5. Gabriel phthalimide synthesis
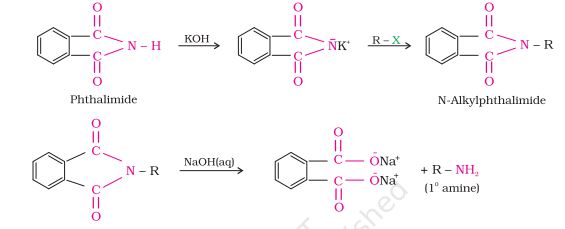
6. Hoffmann bromamide degradation reaction
Hoffmann developed a method for preparation of primary amines by treating an amide with bromine in an aqueous or ethanolic solution of sodium hydroxide. In this degradation reaction, migration of an alkyl or aryl group takes place from carbonyl carbon of the amide to the nitrogen atom. The amine so formed contains one carbon less than that present in the amide.
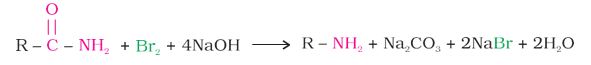
EXAMPLE 2
Write chemical equations for the following conversions:
1. CH3–CH2–Cl into CH3–CH2–CH2–NH2
2. C6H5–CH2–Cl into C6H5–CH2–CH2–NH2
SOLUTION
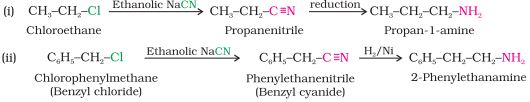
EXAMPLE 3
Write structures and IUPAC names of1. the amide which gives propanamine by Hoffmann bromamide reaction.
2. the amine produced by the Hoffmann degradation of benzamide.
SOLUTION
1. Propanamine contains three carbons. Hence, the amide molecule must contain four carbon atoms. Structure and IUPAC name of the starting amide with four carbon atoms are given below:,,

2. Benzamide is an aromatic amide containing seven carbon atoms. Hence, the amine formed from benzamide is aromatic primary amine containing six carbon atoms.

INTEXT QUESTION
How will you convert
(1) Benzene into aniline (ii) Benzene into N, N-dimethylaniline
(iii) Cl–(CH2)4–Cl into hexan-1,6-diamine?
Physical Properties
The lower aliphatic amines are gases with fishy odour. Primary amines with three or more carbon atoms are liquid and still higher ones are solid. Aniline and other arylamines are usually colourless but get coloured on storage due to atmospheric oxidation.
Lower aliphatic amines are soluble in water because they can form hydrogen bonds with water molecules. However, solubility decreases with increase in molar mass of amines due to increase in size of the hydrophobic alkyl part. Higher amines are essentially insoluble in water. Considering the electronegativity of nitrogen of amine and oxygen of alcohol as 3.0 and 3.5 respectively, you can predict the pattern of solubility of amines and alcohols in water. Out of butan-1-ol and butan-1-amine, which will be more soluble in water and why? Amines are soluble in organic solvents like alcohol, ether and benzene. You may remember that alcohols are more polar than amines and form stronger intermolecular hydrogen bonds than amines.
Primary and secondary amines are engaged in intermolecular association due to hydrogen bonding between nitrogen of one and hydrogen of another molecule. This intermolecular association is more in primary amines than in secondary amines as there are two hydrogen atoms available for hydrogen bond formation in it. Tertiary amines do not have intermolecular association due to the absence of hydrogen atom available for hydrogen bond formation. Therefore, the order of boiling points of isomeric amines is as follows:
Primary > Secondary > Tertiary
Intermolecular hydrogen bonding in primary amines is shown in Fig. 13.2.
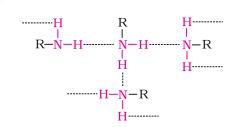
Fig. 13.2 Intermolecular hydrogen bonding in primary amines
Boiling points of amines, alcohols and alkanes of almost the same molar mass are shown in Table 13.2.
Table 13.2: Comparison of Boiling Points of Amines, Alcohols and Alkanes of Similar Molecular Masses

Chemical Reactions
Difference in electronegativity between nitrogen and hydrogen atoms and the presence of unshared pair of electrons over the nitrogen atom makes amines reactive. The number of hydrogen atoms attached to nitrogen atom also decides the course of reaction of amines; that is why primary (–NH2), secondary
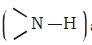 and tertiary amines
and tertiary amines 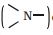 differ in many reactions. Moreover, amines behave as nucleophiles due to the presence of unshared electron pair. Some of the reactions of amines are described below:
differ in many reactions. Moreover, amines behave as nucleophiles due to the presence of unshared electron pair. Some of the reactions of amines are described below:1.Basic character of amines
Amines, being basic in nature, react with acids to form salts.
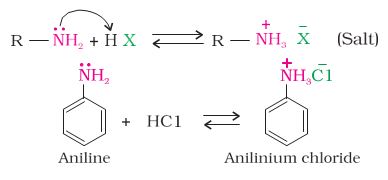
Amine salts on treatment with a base like NaOH, regenerate the parent amine.
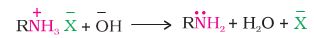
Amine salts are soluble in water but insoluble in organic solvents like ether. This reaction is the basis for the separation of amines from the non basic organic compounds insoluble in water.
The reaction of amines with mineral acids to form ammonium salts shows that these are basic in nature. Amines have an unshared pair of electrons on nitrogen atom due to which they behave as Lewis base. Basic character of amines can be better understood in terms of their Kb and pKb values as explained below:
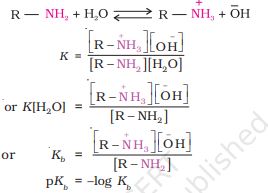
Larger the value of Kb or smaller the value of pKb, stronger is the base. The pKb values of few amines are given in Table 13.3.
Table 13.3: pKb Values of Amines in Aqueous Phase

pKb value of ammonia is 4.75. Aliphatic amines are stronger bases than ammonia due to +I effect of alkyl groups leading to high electron density on the nitrogen atom. Their pKb values lie in the range of 3 to 4.22. On the other hand, aromatic amines are weaker bases than ammonia due to the electron withdrawing nature of the aryl group.
You may find some discrepancies while trying to interpret the Kb values of amines on the basis of +I or –I effect of the substituents present in amines. Besides inductive effect, there are other effects like solvation effect, steric hinderance, etc., which affect the basic strength of amines. Just ponder over. You may get the answer in the following paragraphs.
(a) Alkanamines versus ammonia
Let us consider the reaction of an alkanamine and ammonia with a proton to compare their basicity.
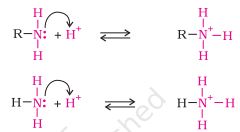
Due to the electron releasing nature of alkyl group, it (R) pushes electrons towards nitrogen and thus makes the unshared electron pair more available for sharing with the proton of the acid. Moreover, the substituted ammonium ion formed from the amine gets stabilised due to dispersal of the positive charge by the +I effect of the alkyl group. Hence, alkylamines are stronger bases than ammonia. Thus, the basic nature of aliphatic amines should increase with increase in the number of alkyl groups. This trend is followed in the gaseous phase. The order of basicity of amines in the gaseous phase follows the expected order: tertiary amine > secondary amine > primary amine > NH3. The trend is not regular in the aqueous state as evident by their pKb values given in Table 13.3. In the aqueous phase, the substituted ammonium cations get stabilised not only by electron releasing effect of the alkyl group (+I) but also by solvation with water molecules. The greater the size of the ion, lesser will be the solvation and the less stabilised is the ion. The order of stability of ions are as follows:
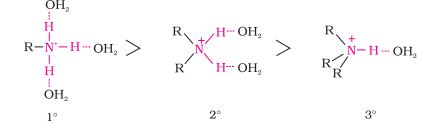
Decreasing order of extent of H-bonding in water and order of stability of ions by solvation.
Greater is the stability of the substituted ammonium cation, stronger should be the corresponding amine as a base. Thus, the order of basicity of aliphatic amines should be: primary > secondary > tertiary, which is opposite to the inductive effect based order. Secondly, when the alkyl group is small, like –CH3 group, there is no steric hindrance to H-bonding. In case the alkyl group is bigger than CH3 group, there will be steric hinderance to H-bonding. Therefore, the change of nature of the alkyl group, e.g., from –CH3 to –C2H5 results in change of the order of basic strength. Thus, there is a subtle interplay of the inductive effect, solvation effect and steric hinderance of the alkyl group which decides the basic strength of alkyl amines in the aqueous state. The order of basic strength in case of methyl substituted amines and ethyl substituted amines in aqueous solution is as follows:
(C2H5)2NH > (C2H5)3N > C2H5NH2 > NH3
(CH3)2NH > CH3NH2 > (CH3)3N > NH3
(b) Arylamines versus ammonia
pKb value of aniline is quite high. Why is it so? It is because in aniline or other arylamines, the -NH2 group is attached directly to the benzene ring. It results in the unshared electron pair on nitrogen atom to be in conjugation with the benzene ring and thus making it less available for protonation. If you write different resonating structures of aniline, you will find that aniline is a resonance hybrid of the following five structures.
Structure-basicity relationship of amines
Basicity of amines is related to their structure. Basic character of an amine depends upon the ease of formation of the cation by accepting a proton from the acid. The more stable the cation is relative to the amine, more basic is the amine.
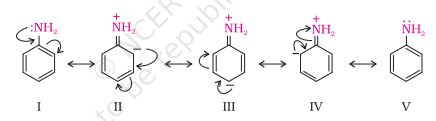
On the other hand, anilinium ion obtained by accepting a proton can have only two resonating structures (kekule).
We know that greater the number of resonating structures, greater is the stability. Thus you can infer that aniline (five resonating structures) is more stable than anilinium ion. Hence, the proton acceptability or the basic nature of aniline or other aromatic amines would be less than that of ammonia. In case of substituted aniline, it is observed that electron releasing groups like –OCH3, –CH3 increase basic strength whereas electron withdrawing groups like –NO2, –SO3H, –COOH, –X decrease it.
EXAMPLE 4
Arrange the following in decreasing order of their basic strength: C6H5NH2, C2H5NH2, (C2H5)2NH, NH3
SOLUTION
The decreasing order of basic strength of the above amines and ammonia follows the following order:
(C2H5)2NH > C2H5NH2 > NH3 > C6H5NH2
2.Alkylation
Amines undergo alkylation on reaction with alkyl halides (refer Unit 10, Class XII).
3. Acylation
Aliphatic and aromatic primary and secondary amines react with acid chlorides, anhydrides and esters by nucleophilic substitution reaction. This reaction is known as acylation. You can consider this reaction as the replacement of hydrogen atom of –NH2 or >N–H group by the acyl group. The products obtained by acylation reaction are known as amides. The reaction is carried out in the presence ofa base stronger than the amine, like pyridine, which removes HCl so formed and shifts the equilibrium to the right hand side.

What do you think is the product of the reaction of amines with carboxylic acids ? They form salts with amines at room temperature.
4. Carbylamine reaction
Aliphatic and aromatic primary amines on heating with chloroform and ethanolic potassium hydroxide form isocyanides or carbylamines which are foul smelling substances. Secondary and tertiary amines do not show this reaction. This reaction is known as carbylamine reaction or isocyanide test and is used as a test for primary amines.
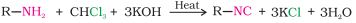
5. Reaction with nitrous acid
Three classes of amines react differently with nitrous acid which is prepared in situ from a mineral acid and sodium nitrite.
(a) Primary aliphatic amines react with nitrous acid to form aliphatic diazonium salts which being unstable, liberate nitrogen gas quantitatively and alcohols. Quantitative evolution of nitrogen is used in estimation of amino acids and proteins.
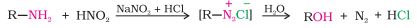
(b) Aromatic amines react with nitrous acid at low temperatures (273-278 K) to form diazonium salts, a very important class of compounds used for synthesis of a variety of aromatic compounds discussed in Section 13.7.
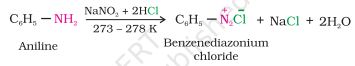
Secondary and tertiary amines react with nitrous acid in a different manner.
6. Reaction with arylsulphonyl chloride
Benzenesulphonyl chloride (C6H5SO2Cl), which is also known as Hinsberg’s reagent, reacts with primary and secondary amines to form sulphonamides.
(a) The reaction of benzenesulphonyl chloride with primary amine yields N-ethylbenzenesulphonyl amide.
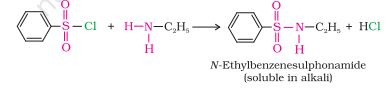
The hydrogen attached to nitrogen in sulphonamide is strongly acidic due to the presence of strong electron withdrawing sulphonyl group. Hence, it is soluble in alkali.
(b) In the reaction with secondary amine, N,N-diethyl- benzenesulphonamide is formed.
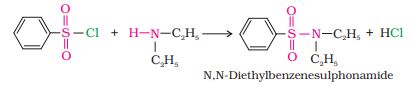
Since N, N-diethylbenzene sulphonamide does not contain any hydrogen atom attached to nitrogen atom, it is not acidic and hence insoluble in alkali.
(c) Tertiary amines do not react with benzenesulphonyl chloride. This property of amines reacting with benzenesulphonyl chloride in a different manner is used for the distinction of primary, secondary and tertiary amines and also for the separation of a mixture of amines. However, these days benzenesulphonyl chloride is replaced by p-toluenesulphonyl chloride.
7. Electrophilic substitution
You have read earlier that aniline is a resonance hybrid of five structures. Where do you find the maximum electron density in these structures? Ortho- and para-positions to the –NH2 group become centres of high electron density. Thus –NH2 group is ortho and para directing and a powerful activating group.
(a) Bromination: Aniline reacts with bromine water at room temperature to give a white precipitate of 2,4,6-tribromoaniline.
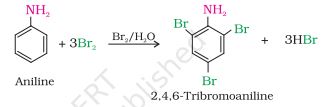
The main problem encountered during electrophilic substitution reactions of aromatic amines is that of their very high reactivity. Substitution tends to occur at ortho- and para-positions. If we have to prepare monosubstituted aniline derivative, how can the activating effect of –NH2 group be controlled ? This can be done by protecting the -NH2 group by acetylation with aceticanhydride, then carrying out the desired substitution followedby hydrolysis of the substituted amide to the substituted amine.
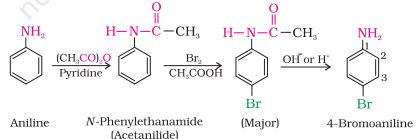
The lone pair of electrons on nitrogen of acetanilide interacts with oxygen atom due to resonance as shown below:
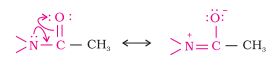
Hence, the lone pair of electrons on nitrogen is less available for donation to benzene ring by resonance. Therefore, activating effect of –NHCOCH3 group is less than that of amino group.(b) Nitration: Direct nitration of aniline yields tarry oxidation products in addition to the nitro derivatives. Moreover, in the strongly acidic medium, aniline is protonated to form the anilinium ion which is meta directing. That is why besides the ortho and para derivatives, significant amount of meta derivative is also formed.
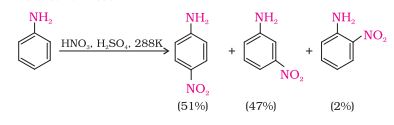
(c) Sulphonation: Aniline reacts with concentrated sulphuric acid to form anilinium hydrogensulphate which on heating with sulphuric acid at 453-473K produces p-aminobenzene sulphonic acid, commonly known as sulphanilic acid, as the major product.
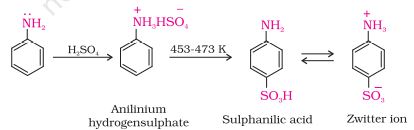
Aniline does not undergo Friedel-Crafts reaction (alkylation and acetylation) due to salt formation with aluminium chloride, the Lewis acid, which is used as a catalyst. Due to this, nitrogen of aniline acquires positive charge and hence acts as a strong deactivating group for further reaction.
INTEXT QUESTION
Arrange the following in increasing order of their basic strength:
1. C2H5NH2, C6H5NH2, NH3, C6H5CH2NH2 and (C2H5)2NH
2. C2H5NH2, (C2H5)2NH, (C2H5)3N, C6H5NH2
3. CH3NH2, (CH3)2NH, (CH3)3N, C6H5NH2, C6H5CH2NH2.
Complete the following acid-base reactions and name the products:
(i) CH3CH2CH2NH2 + HCl \(\to\) (ii) (C2H5)3N + HCl \(\to\)
Write reactions of the final alkylation product of aniline with excess of methyl iodide in the presence of sodium carbonate solution.
Write chemical reaction of aniline with benzoyl chloride and write the name of the product obtained.
Write structures of different isomers corresponding to the molecular formula, C3H9N. IUPAC names of the isomers which will liberate nitrogen gas on treatment with nitrous acid.
DIAZONIUM SALTS
The diazonium salts have the general formula \(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGaamOuamaaxa % cabaGaamOtaaWcbeqaaiabgUcaRaaakmaaBaaaleaacaaIYaaabeaa % kmaanaaabaGaamiwaaaaaaa!3AB5! R{\mathop N\limits^ + _2}\overline X \) where R stands for an aryl group and \(\bar X\) ion may be Cl– Br,– HSO\(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaWaa0baaSqaai % aaisdaaeaacqGHsislaaaaaa!37CE! _4^ - \), BF\(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaWaa0baaSqaai % aaisdaaeaacqGHsislaaaaaa!37CE! _4^ - \) , etc. They are
named by suffixing diazonium to the name of the parent hydrocarbon from which they are formed, followed by the name of anion such as chloride, hydrogensulphate, etc. The \(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaWaaCbiaeaaca % WGobaaleqabaGaey4kaScaaOWaaSbaaSqaaiaaikdaaeqaaaaa!38E6! {\mathop N\limits^ + _2}\) group is called diazonium group. For example, \(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGaam4qamaaBa % aaleaacaaI2aaabeaakiaadIeadaWgaaWcbaGaaGynaaqabaGcdaWf % Gaqaaiaad6eaaSqabeaacqGHRaWkaaGcdaWgaaWcbaGaaGOmaaqaba % GcdaqdaaqaaiaadoeaaaGaamiBaaaa!3E3A! {C_6}{H_5}{\mathop N\limits^ + _2}\overline C l\) is named as benzenediazoniumchloride and \(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGaam4qamaaBa % aaleaacaaI2aaabeaakiaadIeadaWgaaWcbaGaaGynaaqabaGccaWG % obWaa0baaSqaaiaaikdaaeaacqGHRaWkaaGccaWGibGaam4uaiaad+ % eadaqhaaWcbaGaaGinaaqaaiabgkHiTaaaaaa!406F! {C_6}{H_5}N_2^ + HSO_4^ - \) is known as benzenediazonium hydrogensulphate.Primary aliphatic amines form highly unstable alkyldiazonium salts (refer to Section 13.6). Primary aromatic amines form arenediazonium salts which are stable for a short time in solution at low temperatures (273-278 K). The stability of arenediazonium ion is explained on the basis of resonance.

Method OF Preparation of Diazoniun Salts
Benzenediazonium chloride is prepared by the reaction of aniline with nitrous acid at 273-278K. Nitrous acid is produced in the reaction mixture by the reaction of sodium nitrite with hydrochloric acid. The conversion of primary aromatic amines into diazonium salts is known as diazotisation. Due to its instability, the diazonium salt is not generally stored and is used immediately after its preparation.
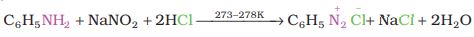
Physical Properties
Benzenediazonium chloride is a colourless crystalline solid. It is readily soluble in water and is stable in cold but reacts with water when warmed. It decomposes easily in the dry state. Benzenediazonium fluoroborate is water insoluble and stable at room temperature.Chemical Reactions
The reactions of diazonium salts can be broadly divided into two categories, namely (A) reactions involving displacement of nitrogen and
(B) reactions involving retention of diazo group.
A. Reactions involving displacement of nitrogen
Diazonium group being a very good leaving group, is substituted by other groups such as Cl–, Br– I– CN– and OH– which displace nitrogen from the aromatic ring. The nitrogen formed escapes from the reaction mixture as a gas.
1. Replacement by halide or cyanide ion: The Cl–, Br– and CN– nucleophiles can easily be introduced in the benzene ring in the presence of Cu(I) ion. This reaction is called Sandmeyer reaction.
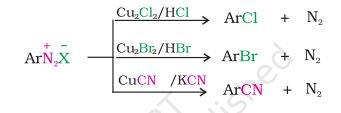
Alternatively, chlorine or bromine can also be introduced in the benzene ring by treating the diazonium salt solution with corresponding halogen acid in the presence of copper powder. This is referred as Gatterman reaction.
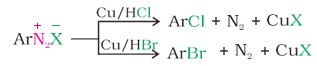
The yield in Sandmeyer reaction is found to be better than Gattermann reaction.
2. Replacement by iodide ion: Iodine is not easily introduced into the benzene ring directly, but, when the diazonium salt solution is treated with potassium iodide, iodobenzene is formed.
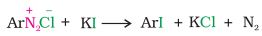

4. Replacement by H : Certain mild reducing agents like hypophosphorous acid (phosphinic acid) or ethanol reduce diazonium salts to arenes and themselves get oxidised to phosphorous acid and ethanal, respectively.
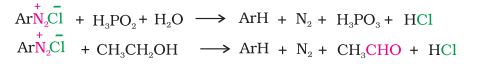
5. Replacement by hydroxyl group: If the temperature of the diazonium salt solution is allowed to rise upto 283 K, the salt gets hydrolysed to phenol.
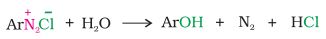
6. Replacement by –NO2 group: When diazonium fluoroborate is heated with aqueous sodium nitrite solution in the presence of copper, the diazonium group is replaced by –NO2 group.

B. Reactions involving retention of diazo group coupling reactions
The azo products obtained have an extended conjugate system having both the aromatic rings joined through the –N=N– bond. These compounds are often coloured and are used as dyes. Benzene diazonium chloride reacts with phenol in which the phenol molecule at its para position is coupled with the diazonium salt to form p-hydroxyazobenzene. This type of reaction is known as coupling reaction. Similarly the reaction of diazonium salt with aniline yields p-aminoazobenzene. This is an example of electrophilic substitution reaction.
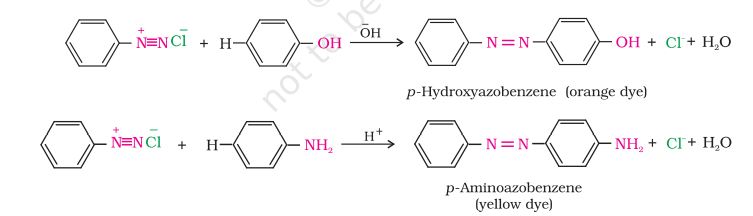
Importance
From the above reactions, it is clear that the diazonium salts are very good intermediates for the introduction of –F, –Cl, –Br, –I, –CN, –OH,
–NO2 groups into the aromatic ring.
Aryl fluorides and iodides cannot be prepared by direct halogenation. The cyano group cannot be introduced by nucleophilic substitution of chlorine in chlorobenzene but cyanobenzene can be easily obtained from diazonium salt.
Thus, the replacement of diazo group by other groups is helpful in preparing those substituted aromatic compounds which cannot be prepared by direct substitution in benzene or substituted benzene.
EXAMPLE 5
How will you convert 4-nitrotoluene to 2-bromobenzoic acid ?
SOLUTION
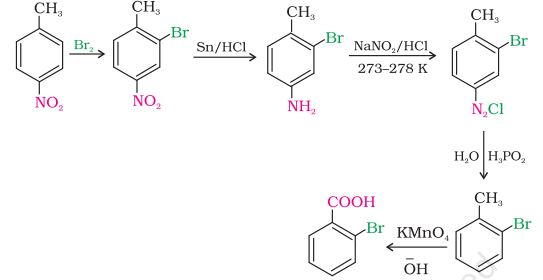
INTEXT QUESTION
Convert
1. 3-Methylaniline into 3-nitrotoluene.
2.Aniline into 1,3,5 - tribromobenzene.
-
Biomolecules.14
A living system grows, sustains and reproduces itself. The most amazing thing about a living system is that it is composed of non-living atoms and molecules. The pursuit of knowledge of what goes on chemically within a living system falls in the domain of biochemistry. Living systems are made up of various complex biomolecules like carbohydrates, proteins, nucleic acids, lipids, etc. Proteins and carbohydrates are essential constituents of our food. These biomolecules interact with each other and constitute the molecular logic of life processes. In addition, some simple molecules like vitamins and mineral salts also play an important role in the functions of organisms. Structures and functions of some of these biomolecules are discussed in this Unit.
Carbohydrates
Carbohydrates are primarily produced by plants and form a very large group of naturally occurring organic compounds. Some common examples of carbohydrates are cane sugar, glucose, starch, etc. Most of them have a general formula, Cx(H2O)y, and were considered as hydrates of carbon from where the name carbohydrate was derived. For example, the molecular formula of glucose (C6H12O6) fits into this general formula, C6(H2O)6. But all the compounds which fit into this formula may not be classified as carbohydrates. For example acetic acid (CH3COOH) fits into this general formula, C2(H2O)2 but is not a carbohydrate. Similarly, rhamnose, C6H12O5 is a carbohydrate but does not fit in this definition. A large number of their reactions have shown that they contain specific functional groups. Chemically, the carbohydrates may be defined as optically active polyhydroxy aldehydes or ketones or the compounds which produce such units on hydrolysis. Some of the carbohydrates, which are sweet in taste, are also called sugars. The most common sugar, used in our homes is named as sucrose whereas the sugar present in milk is known as lactose. Carbohydrates are also called saccharides (Greek: sakcharon means sugar).
Classification of Carbohydrates
Carbohydrates are classified on the basis of their behaviour on hydrolysis. They have been broadly divided into following three groups.
1. Monosaccharides: A carbohydrate that cannot be hydrolysed further to give simpler unit of polyhydroxy aldehyde or ketone is called a monosaccharide. About 20 monosaccharides are known to occur in nature. Some common examples are glucose, fructose, ribose, etc.
2. Oligosaccharides: Carbohydrates that yield two to ten monosaccharide units, on hydrolysis, are called oligosaccharides. They are further classified as disaccharides, trisaccharides, tetrasaccharides, etc., depending upon the number of monosaccharides, they provide on hydrolysis. Amongst these the most common are disaccharides. The two monosaccharide units obtained on hydrolysis of a disaccharide may be same or different. For example, one molecule of sucrose on hydrolysis gives one molecule of glucose and one molecule of fructose whereas maltose gives two molecules of only glucose.
3.Polysaccharides: Carbohydrates which yield a large number of monosaccharide units on hydrolysis are called polysaccharides. Some common examples are starch, cellulose, glycogen, gums, etc. Polysaccharides are not sweet in taste, hence they are also called non-sugars.
The carbohydrates may also be classified as either reducing or non- reducing sugars. All those carbohydrates which reduce Fehling’s solution and Tollens’ reagent are referred to as reducing sugars. All monosaccharides whether aldose or ketose are reducing sugars.
Monosaccharides
Monosaccharides are further classified on the basis of number of carbon atoms and the functional group present in them. If a monosaccharide contains an aldehyde group, it is known as an aldose and if it contains a keto group, it is known as a ketose. Number of carbon atoms constituting the monosaccharide is also introduced in the name as is evident from the examples given in Table 14.1
Table 14.1: Different Types of Monosaccharides
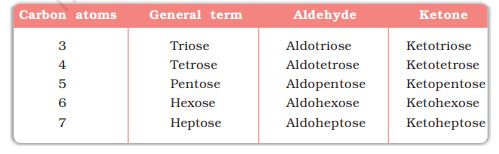
Glucose
Glucose occurs freely in nature as well as in the combined form. It is present in sweet fruits and honey. Ripe grapes also contain glucose in large amounts. It is prepared as follows:
Preparation OF Glucose
1. From sucrose (Cane sugar): If sucrose is boiled with dilute HCl or H2SO4 in alcoholic solution, glucose and fructose are obtained in equal amounts.
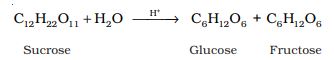
2. From starch: Commercially glucose is obtained by hydrolysis of starch by boiling it with dilute H2SO4 at 393 K under pressure.
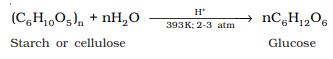
Structure OF Glucose
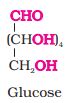
Glucose is an aldohexose and is also known as dextrose. It is the monomer of many of the larger carbohydrates, namely starch, cellulose. It is probably the most abundant organic compound on earth. It was assigned the structure given below on the basis of the following evidences:
1. Its molecular formula was found to be C6H12O6.
2. On prolonged heating with HI, it forms n-hexane, suggesting that all the six carbon atoms are linked in a straight chain.
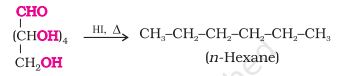
3. Glucose reacts with hydroxylamine to form an oxime and adds a molecule of hydrogen cyanide to give cyanohydrin. These reactions confirm the presence of a carbonyl group (>C = O) in glucose.
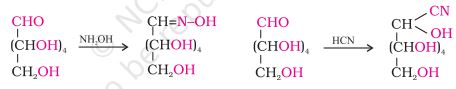
4. Glucose gets oxidised to six carbon carboxylic acid (gluconic acid) on reaction with a mild oxidising agent like bromine water. This indicates that the carbonyl group is present as an aldehydic group.
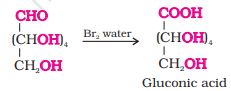
5. Acetylation of glucose with acetic anhydride gives glucose pentaacetate which confirms the presence of five –OH groups. Since it exists as a stable compound, five –OH groups should be attached to different carbon atoms.
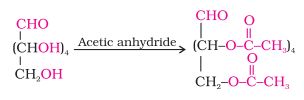
6. On oxidation with nitric acid, glucose as well as gluconic acid both yield a dicarboxylic acid, saccharic acid. This indicates the presence of a primary alcoholic (–OH) group in glucose.
The exact spatial arrangement of different —OH groups was given by Fischer after studying many other properties. Its configuration is correctly represented as I. So gluconic acid is represented as II and saccharic acid as III.
Glucose is correctly named as D(+)-glucose. ‘D’ before the name of glucose represents the configuration whereas ‘(+)’ represents dextrorotatory nature of the molecule. It should be remembered that ‘D’ and ‘L’ have no relation with the optical activity of the compound. They are also not related to letter ‘d’ and ‘l’ (see Unit 10). The meaning of D– and L– notations is as follows.
The letters ‘D’ or ‘L’ before the name of any compound indicate the relative configuration of a particular stereoisomer of a compound with respect to configuration of some other compound, configuration of which is known. In the case of carbohydrates, this refers to their relation with a particular isomer of glyceraldehyde. Glyceraldehyde contains one asymmetric carbon atom and exists in two enantiomeric forms as shown below.
(+) Isomer of glyceraldehyde has ‘D’ configuration. It means that when its structural formula is written on paper following specific conventions which you will study in higher classes, the –OH group lies on right hand side in the structure. All those compounds which can be chemically correlated to D (+) isomer of glyceraldehyde are said to have D- configuration whereas those which can be correlated to ‘L’ (–) isomer of glyceraldehyde are said to have L—configuration. In L (–) isomer –OH group is on left hand side as you can see in the structure. For assigning the configuration of monosaccharides, it is the lowest asymmetric carbon atom (as shown below) which is compared. As in (+) glucose, —OH on the lowest asymmetric carbon is on the right side which is comparable to (+) glyceraldehyde, so (+) glucose is assigned D-configuration. Other asymmetric carbon atoms of glucose are not considered for this comparison. Also, the structure of glucose and glyceraldehyde is written in a way that most oxidised carbon (in this case –CHO)is at the top.

Cyclic Structure of Glucose
The structure (I) of glucose explained most of its properties but the following reactions and facts could not be explained by this structure.
1. Despite having the aldehyde group, glucose does not give Schiff’s test and it does not form the hydrogensulphite addition product with NaHSO3.
2. The pentaacetate of glucose does not react with hydroxylamine indicating the absence of free —CHO group.
3. Glucose is found to exist in two different crystalline forms which are named as \(\alpha\) and \(\beta \). The \(\alpha\)-form of glucose (m.p. 419 K) is obtained by crystallisation from concentrated solution of glucose at 303 K while the \(\beta \)-form (m.p. 423 K) is obtained by crystallisation from hot and saturated aqueous solution at 371 K.
This behaviour could not be explained by the open chain structure
(I) for glucose. It was proposed that one of the —OH groups may add to the —CHO group and form a cyclic hemiacetal structure. It was found that glucose forms a six-membered ring in which —OH at C-5 is involved in ring formation. This explains the absence of —CHO group and also existence of glucose in two forms as shown below. These two cyclic forms exist in equilibrium with open chain structure.
The two cyclic hemiacetal forms of glucose differ only in the configuration of the hydroxyl group at C1, called anomeric carbon
(the aldehyde carbon before cyclisation). Such isomers, i.e., \(\alpha\)-form and \(\beta\)-form, are called anomers. The six membered cyclic structure of glucose is called pyranose structure (\(\alpha\)– or \(\beta\)–), in analogy with pyran. Pyran is a cyclic organic compound with one oxygen atom and five carbon atoms in the ring. The cyclic structure of glucose is more correctly represented by Haworth structure as given below.
Fructose
Fructose is an important ketohexose. It is obtained along with glucose by the hydrolysis of disaccharide, sucrose. It is a natural monosaccharide found in fruits, honey and vegetables. In its pure form it is used as a sweetner. It is also an important ketohexose.
Structure of Fructose
Fructose also has the molecular formula C6H12O6 and on the basis of its reactions it was found to contain a ketonic functional group at carbon number 2 and six carbons in straight chain as in the case of glucose. It belongs to D-series and is a laevorotatory compound. It is appropriately written as D-(–)-fructose. Its open chain structure is as shown.
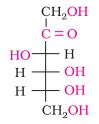
It also exists in two cyclic forms which are obtained by the addition of —OH at C5 to the
 group. The ring, thus formed is a five membered ring and is named as furanose with analogy to the compound furan. Furan is a five membered cyclic compound with one oxygen and four carbon atoms.
group. The ring, thus formed is a five membered ring and is named as furanose with analogy to the compound furan. Furan is a five membered cyclic compound with one oxygen and four carbon atoms.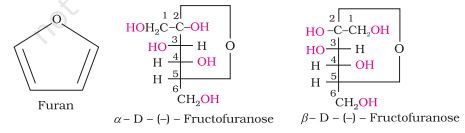
The cyclic structures of two anomers of fructose are represented by Haworth structures as given.
Disaccharides
You have already read that disaccharides on hydrolysis with dilute acids or enzymes yield two molecules of either the same or different monosaccharides. The two monosaccharides are joined together by an oxide linkage formed by the loss of a water molecule. Such a linkage between two monosaccharide units through oxygen atom is called glycosidic linkage.
In disaccharides, if the reducing groups of monosaccharides i.e., aldehydic or ketonic groups are bonded, these are non-reducing sugars, e.g., sucrose. On the other hand, sugars in which these functional groups are free, are called reducing sugars, for example, maltose and lactose.
1. Sucrose: One of the common disaccharides is sucrose which on hydrolysis gives equimolar mixture of D-(+)-glucose and D-(-) fructose.
These two monosaccharides are held together by a glycosidic linkage between C1 of \(\alpha\)-D-glucose and C2 of \(\beta\)-D-fructose. Since the reducing groups of glucose and fructose are involved in glycosidic bond formation, sucrose is a non reducing sugar.
Sucrose
Sucrose is dextrorotatory but after hydrolysis gives dextrorotatory glucose and laevorotatory fructose. Since the laevorotation of fructose (–92.4°) is more than dextrorotation of glucose (+ 52.5°), the mixture is laevorotatory. Thus, hydrolysis of sucrose brings about a change in the sign of rotation, from dextro (+) to laevo (–) and the product is named as invert sugar.
(2) Maltose: Another disaccharide, maltose is composed of two \( \alpha\)-D-glucose units in which C1 of one glucose (I) is linked to C4 of another glucose unit (II). The free aldehyde group can be produced at C1 of second glucose in solution and it shows reducing properties so it is a reducing sugar.
Maltose
(3) Lactose: It is more commonly known as milk sugar since this disaccharide is found in milk. It is composed of \(\beta\)-D-galactose and \(\beta\)-D-glucose. The linkage is between C1 of galactose and C4 of glucose. Free aldehyde group may be produced at C-1 of glucose unit, hence it is also a reducing sugar.
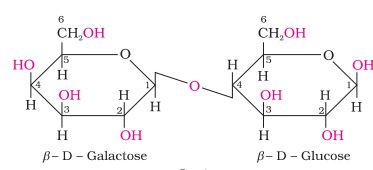
Lactose
Polysaccharides
Polysaccharides contain a large number of monosaccharide units joined together by glycosidic linkages. These are the most commonly encountered carbohydrates in nature. They mainly act as the food storage or structural materials.
1. Starch: Starch is the main storage polysaccharide of plants. It is the most important dietary source for human beings. High content of starch is found in cereals, roots, tubers and some vegetables. It is a polymer of \( \alpha\)-glucose and consists of two components— Amylose and Amylopectin. Amylose is water soluble component which constitutes about 15-20% of starch. Chemically amylose is a long unbranched chain with 200-1000 -D-(+)-glucose units held together by C1– C4 glycosidic linkage.
Amylopectin is insoluble in water and constitutes about 80- 85% of starch. It is a branched chain polymer of \( \alpha\)-D-glucose units in which chain is formed by C1–C4 glycosidic linkage whereas branching occurs by C1–C6 glycosidic linkage.
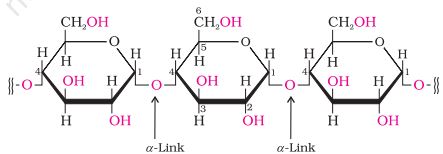
Amylose
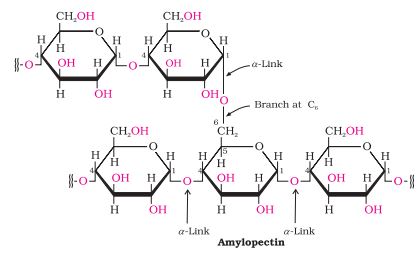
(2) Cellulose: Cellulose occurs exclusively in plants and it is the most abundant organic substance in plant kingdom. It is a predominant constituent of cell wall of plant cells. Cellulose is a straight chain
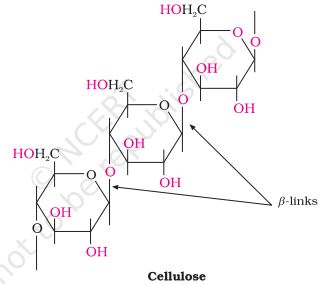
polysaccharide composed only of \(\beta\)-D-glucose units which are joined by glycosidic linkage between C1 of one glucose unit and C4 of the next glucose unit.
(3) Glycogen: The carbohydrates are stored in animal body as glycogen. It is also known as animal starch because its structure is similar to amylopectin and is rather more highly branched. It is present in liver, muscles and brain. When the body needs glucose, enzymes break the glycogen down to glucose. Glycogen is also found in yeast and fungi.
Importance of Carbohydrates
Carbohydrates are essential for life in both plants and animals. They form a major portion of our food. Honey has been used for a long time as an instant source of energy by ‘Vaids’ in ayurvedic system of medicine. Carbohydrates are used as storage molecules as starch in plants and glycogen in animals. Cell wall of bacteria and plants is made up of cellulose. We build furniture, etc. from cellulose in the form of wood and clothe ourselves with cellulose in the form of cotton fibre. They provide raw materials for many important industries like textiles, paper, lacquers and breweries.
Two aldopentoses viz. D-ribose and 2-deoxy-D-ribose (Section 14.5.1, Class XII) are present in nucleic acids. Carbohydrates are found in biosystem in combination with many proteins and lipids.
INTEXT QUESTION
Glucose or sucrose are soluble in water but cyclohexane or benzene (simple six membered ring compounds) are insoluble in water. Explain.
What are the expected products of hydrolysis of lactose?
How do you explain the absence of aldehyde group in the pentaacetate of D-glucose?
Proteins
Proteins are the most abundant biomolecules of the living system. Chief sources of proteins are milk, cheese, pulses, peanuts, fish, meat, etc. They occur in every part of the body and form the fundamental basis of structure and functions of life. They are also required for growth and maintenance of body. The word protein is derived from Greek word, “proteios” which means primary or of prime importance. All proteins are polymers of \(\alpha\)-amino acids.
Amino Acids
Amino acids contain amino (–NH2) and carboxyl (–COOH) functional groups. Depending upon the relative position of amino group with respect to carboxyl group, the amino acids can be classified as \(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGaeqySdeMaai % ilaiabek7aIjaacYcacqaHZoWzcaGGSaGaeqiTdqgaaa!3E92! \alpha ,\beta ,\gamma ,\delta \) and so on. Only \(\alpha\)-amino acids are obtained on hydrolysis of proteins. They may contain other functional groups also.
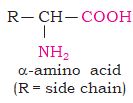
All \(\alpha\)-amino acids have trivial names, which usually reflect the property of that compound or its source. Glycine is so named since it has sweet taste (in Greek glykos means sweet) and tyrosine was first obtained from cheese (in Greek, tyros means cheese.) Amino acids are generally represented by a three letter symbol, sometimes one letter symbol is also used. Structures of some commonly occurring amino acids along with their 3-letter and 1-letter symbols are given in Table 14.2.
Table 14.2: Natural Amino Acids
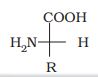
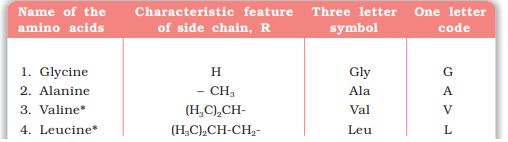
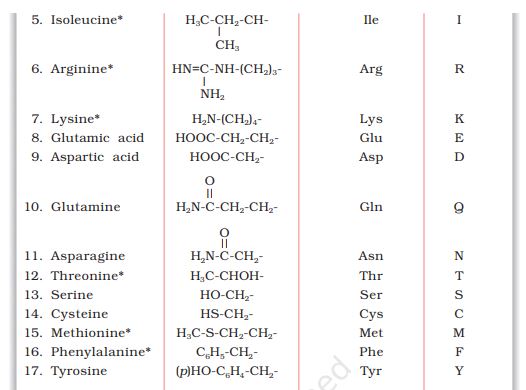
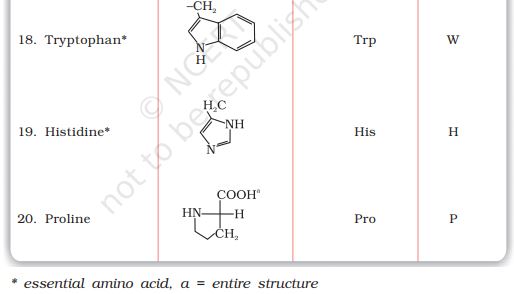
Classification OF Amino Acids
Amino acids are classified as acidic, basic or neutral depending upon the relative number of amino and carboxyl groups in their molecule. Equal number of amino and carboxyl groups makes it neutral; more number of amino than carboxyl groups makes it basic and more carboxyl groups as compared to amino groups makes it acidic. The amino acids, which can be synthesised in the body, are known as non- essential amino acids. On the other hand, those which cannot be synthesised in the body and must be obtained through diet, are known as essential amino acids (marked with asterisk in Table 14.2).
Amino acids are usually colourless, crystalline solids. These are water-soluble, high melting solids and behave like salts rather than simple amines or carboxylic acids. This behaviour is due to the presence of both acidic (carboxyl group) and basic (amino group) groups in the same molecule. In aqueous solution, the carboxyl group can lose a proton and amino group can accept a proton, giving rise to a dipolar ion known as zwitter ion. This is neutral but contains both positive and negative charges.
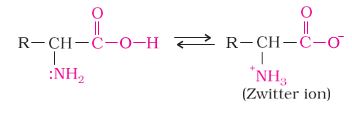
In zwitter ionic form, amino acids show amphoteric behaviour as they react both with acids and bases.
Except glycine, all other naturally occurring \(\alpha\)-amino acids are optically active, since the \(\alpha\)-carbon atom is asymmetric. These exist both in ‘D’ and ‘L’ forms. Most naturally occurring amino acids have L-configuration. L-Aminoacids are represented by writing the –NH2 group on left hand side.
Structure of Proteins
You have already read that proteins are the polymers of \(\alpha\)-amino acids and they are connected to each other by peptide bond or peptide linkage. Chemically, peptide linkage is an amide formed between–COOH group and –NH2 group. The reaction between two molecules of similar or different amino acids, proceeds through the combination of the amino group of one molecule with the carboxyl group of the other. This results in the elimination of a water molecule and formation of a peptide bond –CO–NH–. The product of the reaction is called a dipeptide because it is made up of two amino acids. For example, when carboxyl group of glycine combines with the amino group of alanine we get a dipeptide, glycylalanine.
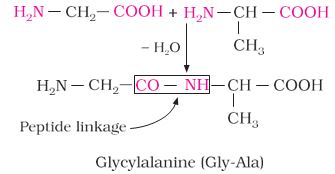
If a third amino acid combines to a dipeptide, the product is called a tripeptide. A tripeptide contains three amino acids linked by two peptide linkages. Similarly when four, five or six amino acids are linked, the respective products are known as tetrapeptide, pentapeptide or hexapeptide, respectively. When the number of such amino acids is more than ten, then the products are called polypeptides. A polypeptide with more than hundred amino acid residues, having molecular mass higher than 10,000u is called a protein. However, the distinction between a polypeptide and a protein is not very sharp. Polypeptides with fewer amino acids are likely to be called proteins if they ordinarily have a well defined conformation of a protein such as insulin which contains 51 amino acids.
Proteins can be classified into two types on the basis of their molecular shape.
(a) Fibrous proteins
When the polypeptide chains run parallel and are held together by hydrogen and disulphide bonds, then fibre– like structure is formed. Such proteins are generally insoluble in water. Some common examples are keratin (present in hair, wool, silk) and myosin (present in muscles), etc.
(b) Globular proteins
This structure results when the chains of polypeptides coil around to give a spherical shape. These are usually soluble in water. Insulin and albumins are the common examples of globular proteins.
Structure and shape of proteins can be studied at four different levels, i.e., primary, secondary, tertiary and quaternary, each level being more complex than the previous one.
(1) Primary structure of proteins: Proteins may have one or more polypeptide chains. Each polypeptide in a protein has amino acids linked with each other in a specific sequence and it is this sequence of amino acids that is said to be the primary structure of that protein. Any change in this primary structure i.e., the sequence of amino acids creates a different protein.
(2) Secondary structure of proteins: The secondary structure of protein refers to the shape in which a long polypeptide chain can exist. They are found to exist in two different types of structures viz. \(\alpha\)-helix and \(\beta\)-pleated sheet structure. These structures arise dueto the regular folding of the backbone of the polypeptide chain due to hydrogen bonding between
 and –NH– groups of the peptide bond.
and –NH– groups of the peptide bond.\(\alpha\)-Helix is one of the most common ways in which a polypeptide chain forms all possible hydrogen bonds by twisting into a right handed screw (helix) with the –NH group of each amino acid residue hydrogen bonded to the
 of an adjacent turn of the helix as shown in Fig.14.1.
of an adjacent turn of the helix as shown in Fig.14.1.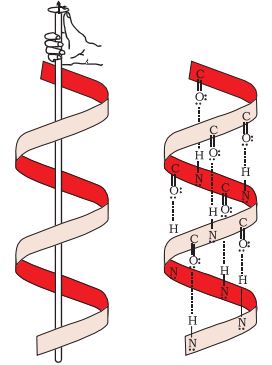
Fig. 14.1: \(\alpha\)-Helix structure of proteins
In \(\beta\)-pleated sheet structure all peptide chains are stretched out to nearly maximum extension and then laid side by side which are held together by intermolecular hydrogen bonds. The structure resembles the pleated folds of drapery and therefore is known as \(\beta\)-pleated sheet.
(3) Tertiary structure of proteins: The tertiary structure of proteins represents overall folding of the polypeptide chains i.e., further folding of the secondary structure. It gives rise to two major molecular shapes viz. fibrous and globular. The main forces which stabilise the 2° and 3° structures of proteins are hydrogen bonds, disulphide linkages, van der Waals and electrostatic forces of attraction.
(4) Quaternary structure of proteins: Some of the proteins are composed of two or more polypeptide chains referred to as sub-units. The spatial arrangement of these subunits with respect to each other is known as quaternary structure.
Fig. 14.2: \(\beta\)-Pleated sheet structure of proteins
A diagrammatic representation of all these four structures is given in Figure 14.3 where each coloured ball represents an amino acid.
Fig. 14.3: Diagrammatic representation of protein structure (two sub-units of two types in quaternary structure)
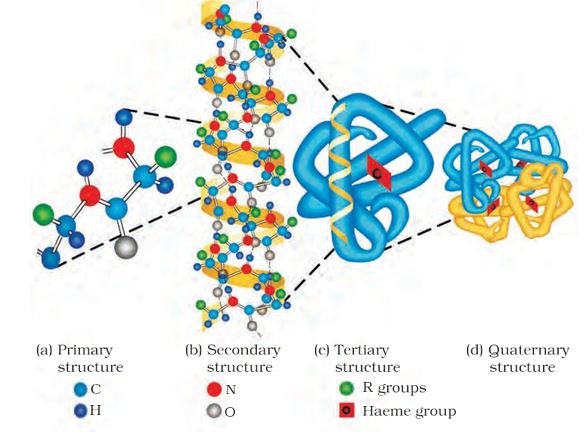
Fig. 14.4: Primary, secondary, tertiary and quaternary structures of haemoglobin
Denaturation OF Proteins
Protein found in a biological system with a unique three-dimensional structure and biological activity is called a native protein. When a protein in its native form, is subjected to physical change like change in temperature or chemical change like change in pH, the hydrogen bonds are disturbed. Due to this, globules unfold and helix get uncoiled and protein loses its biological activity. This is called denaturation of
protein. During denaturation secondary and tertiary structures are destroyed but primary structure remains intact. The coagulation of egg white on boiling is a common example of denaturation. Another example is curdling of milk which is caused due to the formation of lactic acid by the bacteria present in milk.
INTEXT QUESTION
The melting points and solubility in water of amino acids are generally higher than that of the corresponding halo acids. Explain.
Where does the water present in the egg go after boiling the egg?
Enzymes
Life is possible due to the coordination of various chemical reactions in living organisms. An example is the digestion of food, absorption of appropriate molecules and ultimately production of energy. This process involves a sequence of reactions and all these reactions occur in the body under very mild conditions. This occurs with the help of certain biocatalysts called enzymes. Almost all the enzymes are globular proteins. Enzymes are very specific for a particular reaction and for a particular substrate. They are generally named after the compound or class of compounds upon which they work. For example, the enzyme that catalyses hydrolysis of maltose into glucose is named as maltase.
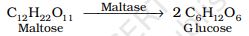
Sometimes enzymes are also named after the reaction, where they are used. For example, the enzymes which catalyse the oxidation of one substrate with simultaneous reduction of another substrate are named as oxidoreductase enzymes. The ending of the name of an enzyme is -ase.
Mechanism of Enzyme Action
Enzymes are needed only in small quantities for the progress of a reaction. Similar to the action of chemical catalysts, enzymes are said to reduce the magnitude of activation energy. For example, activation energy for acid hydrolysis of sucrose is 6.22 kJ mol–1, while the activation energy is only 2.15 kJ mol–1 when hydrolysed by the enzyme, sucrase. Mechanism for the enzyme action has been discussed in Unit 5.4.
Vitamins
It has been observed that certain organic compounds are required in small amounts in our diet but their deficiency causes specific diseases. These compounds are called vitamins. Most of the vitamins cannot be synthesised in our body but plants can synthesise almost all of them, so they are considered as essential food factors. However, the bacteria of the gut can produce some of the vitamins required by us. All the vitamins are generally available in our diet. Different vitamins belong to various chemical classes and it is difficult to define them on the basis of structure. They are generally regarded as organic compounds required in the diet in small amounts to perform specific biological functions for normal maintenance of optimum growth and health of the organism. Vitamins are designated by alphabets A, B, C, D, etc. Some of them are further named as sub-groups e.g. B1, B2, B6, B12, etc. Excess of vitamins is also harmful and vitamin pills should not be taken without the advice of doctor.
The term “Vitamine” was coined from the word vital + amine since the earlier identified compounds had amino groups. Later work showed that most of them did not contain amino groups, so the letter ‘e’ was dropped and the term vitamin is used these days.
Classification OF Vitamins
Vitamins are classified into two groups depending upon their solubility in water or fat.
1. Fat soluble vitamins: Vitamins which are soluble in fat and oils but insoluble in water are kept in this group. These are vitamins A, D, E and K. They are stored in liver and adipose (fat storing) tissues.
2. Water soluble vitamins: B group vitamins and vitamin C are soluble in water so they are grouped together. Water soluble vitamins must be supplied regularly in diet because they are readily excreted in urine and cannot be stored (except vitamin B12) in our body.
Some important vitamins, their sources and diseases caused by their deficiency are listed in Table 14.3.
Table 14.3: Some important Vitamins, their Sources and their Deficiency Diseases
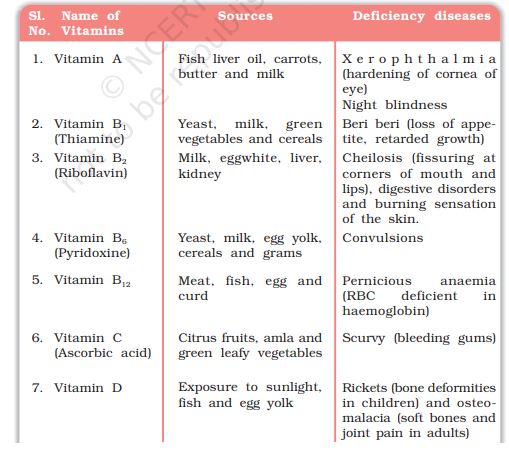

Nucleic Acids
Every generation of each and every species resembles its ancestors in many ways. How are these characteristics transmitted from one generation to the next? It has been observed that nucleus of a living cell is responsible for this transmission of inherent characters, also called heredity. The particles in nucleus of the cell, responsible for heredity, are called chromosomes which are made up of proteins and another type of biomolecules called nucleic acids. These are mainly of two types, the deoxyribonucleic acid (DNA) and ribonucleic acid (RNA). Since nucleic acids are long chain polymers of nucleotides, so they are also called polynucleotides.
James Dewey Watson
Born in Chicago, Illinois, in 1928, Dr Watson received his Ph.D. (1950) from Indiana University in Zoology. He is best known for his discovery of the structure of DNA for which he shared with Francis Crick and Maurice Wilkins the 1962 Nobel prize in Physiology and Medicine. They proposed that DNA molecule takes the shape of a double helix, an elegantly simple structure that resembles a gently twisted ladder. The rails of the ladder are made of alternating units of phosphate and the sugar deoxyribose;
the rungs are each composed of a pair of purine/ pyrimidine bases. This research laid the foundation for the emerging field of molecular biology. The complementary pairing of nucleotide bases explains how identical copies of parental DNA pass on to two daughter cells. This research launched a revolution in biology that led to modern recombinant DNA techniques
Chemical Composition of Nucleic Acids
Complete hydrolysis of DNA (or RNA) yields a pentose sugar, phosphoric acid and nitrogen containing heterocyclic compounds (called bases). In DNA molecules, the sugar moiety is \(\beta\)-D-2-deoxyribose whereas in RNA molecule, it is \(\beta\)-D-ribose.

DNA contains four bases viz. adenine (A), guanine (G), cytosine (C) and thymine (T). RNA also contains four bases, the first three bases are same as in DNA but the fourth one is uracil (U).
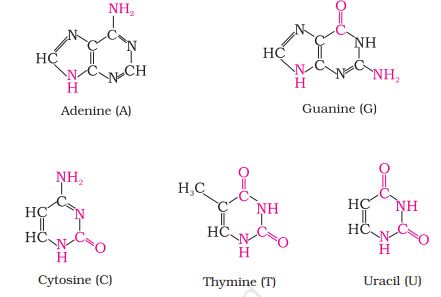
Structure of Nucleic Acids
A unit formed by the attachment of a base to \(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGabGymayaafa % aaaa!36BD! 1'\) position of sugar is known as nucleoside. In nucleosides, the sugar carbons are numbered as \(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGabGymayaafa % GaaiilaiqaikdagaqbaiaacYcaceaIZaGbauaaaaa!39AE! 1',2',3'\) etc. in order to distinguish these from the bases (Fig. 14.5a). When nucleoside is linked to phosphoric acid at \(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGabGymayaafa % aaaa!36BD! 5'\)-position of sugar moiety, we get a nucleotide (Fig. 14.5).
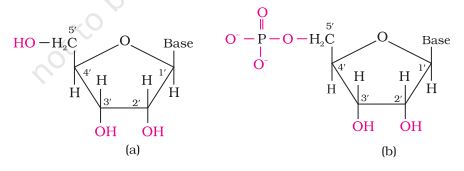
Fig. 14.5: Structure of (a) a nucleoside and (b) a nucleotide
Nucleotides are joined together by phosphodiester linkage between \(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGabGymayaafa % aaaa!36BD! 5'\) and \(% MathType!MTEF!2!1!+- % feaagKart1ev2aaatCvAUfeBSjuyZL2yd9gzLbvyNv2CaerbuLwBLn % hiov2DGi1BTfMBaeXatLxBI9gBaerbd9wDYLwzYbItLDharqqtubsr % 4rNCHbGeaGqiVu0Je9sqqrpepC0xbbL8F4rqqrFfpeea0xe9Lq-Jc9 % vqaqpepm0xbba9pwe9Q8fs0-yqaqpepae9pg0FirpepeKkFr0xfr-x % fr-xb9adbaqaaeGaciGaaiaabeqaamaabaabaaGcbaGabGymayaafa % aaaa!36BD! 3'\) carbon atoms of the pentose sugar. The formation of a typical dinucleotide is shown in Fig. 14.6.
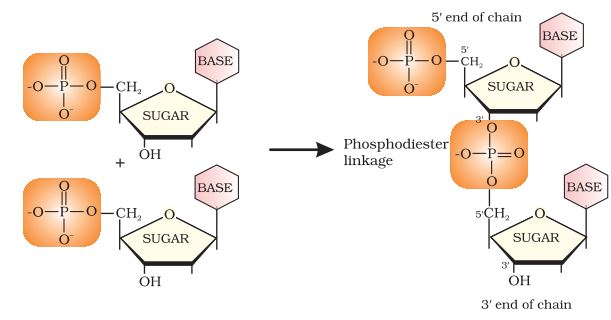
Fig. 14.6: Formation of a dinucleotide
A simplified version of nucleic acid chain is as shown below.
Information regarding the sequence of nucleotides in the chain of a nucleic acid is called its primary structure. Nucleic acids have a secondary structure also. James Watson and Francis Crick gave a double strand helix structure for DNA (Fig. 14.7).
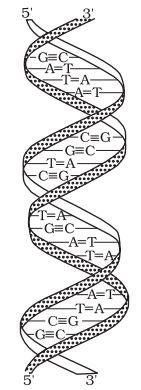
Fig. 14.7: Double strand helix structure for DNA
Two nucleic acid chains are wound about each other and held together by hydrogen bonds between pairs of bases. The two strands are complementary to each other because the hydrogen bonds are formed between specific pairs of bases. Adenine forms hydrogen bonds with thymine whereas cytosine forms hydrogen bonds with guanine.
In secondary structure of RNA single stranded helics is present which sometimes foldsback on itself. RNA molecules are of three types and they perform different functions. They are named as messenger RNA (m-RNA), ribosomal RNA (r-RNA) and transfer RNA (t-RNA).
Har Gobind Khorana
Har Gobind Khorana, was born in 1922. He obtained his M.Sc. degree from Punjab University in Lahore. He worked with Professor Vladimir Prelog, who moulded Khorana’s thought and philosophy towards science, work and effort. After a brief stay in India in 1949, Khorana went back to England and worked with Professor G.W. Kenner and Professor A.R.Todd. It was at Cambridge, U.K. that he got interested in both proteins and nucleic acids. Dr Khorana shared the Nobel Prize for Medicine and Physiology in 1968 with Marshall Nirenberg and Robert Holley for cracking the genetic code.
DNA Fingerprinting
It is known that every individual has unique fingerprints. These occur at the tips of the fingers and have been used for identification for a long time but these can be altered by surgery. A sequence of bases on DNA is also unique for a person and information regarding this is called DNA fingerprinting. It is same for every cell and cannot be altered by any known treatment. DNA fingerprinting is now used
1. in forensic laboratories for identification of criminals.
2. to determine paternity of an individual.
3. to identify the dead bodies in any accident by comparing the DNA’s of parents or children.
4. to identify racial groups to rewrite biological evolution.
Biological Functions of Nucleic Acids
DNA is the chemical basis of heredity and may be regarded as the reserve of genetic information. DNA is exclusively responsible for maintaining the identity of different species of organisms over millions of years. A DNA molecule is capable of self duplication during cell division and identical DNA strands are transferred to daughter cells. Another important function of nucleic acids is the protein synthesis in the cell. Actually, the proteins are synthesised by various RNA molecules in the cell but the message for the synthesis of a particular protein is present in DNA.
Hormones
Hormones are molecules that act as intercellular messengers. These are produced by endocrine glands in the body and are poured directly in the blood stream which transports them to the site of action.
In terms of chemical nature, some of these are steroids, e.g., estrogens and androgens; some are poly peptides for example insulin and endorphins and some others are amino acid derivatives such as epinephrine and norepinephrine.
Hormones have several functions in the body. They help to maintain the balance of biological activities in the body. The role of insulin in keeping the blood glucose level within the narrow limit is an example of this function. Insulin is released in response to the rapid rise in blood glucose level. On the other hand hormone glucagon tends to increase the glucose level in the blood. The two hormones together regulate the glucose level in the blood. Epinephrine and norepinephrine mediate responses to external stimuli. Growth hormones and sex hormones play role in growth and development. Thyroxine produced in the thyroid gland is an iodinated derivative of amino acid tyrosine. Abnormally low level of thyroxine leads to hypothyroidism which is characterised by lethargyness and obesity. Increased level of thyroxine causes hyperthyroidism. Low level of iodine in the diet may lead to hypothyroidism and enlargement of the thyroid gland. This condition is largely being controlled by adding sodium iodide to commercial table salt (“Iodised” salt).
Steroid hormones are produced by adrenal cortex and gonads (testes in males and ovaries in females). Hormones released by the adrenal cortex play very important role in the functions of the body. For example, glucocorticoids control the carbohydrate metabolism, modulate inflammatory reactions, and are involved in reactions to stress. The mineralocorticoids control the level of excretion of water and salt by the kidney. If adrenal cortex does not function properly then one of the results may be Addison’s disease characterised by hypoglycemia, weakness and increased susceptibility to stress. The disease is fatal unless it is treated by glucocorticoids and mineralocorticoids. Hormones released by gonads are responsible for development of secondary sex characters. Testosterone is the major sex hormone produced in males. It is responsible for development of secondary male characteristics (deep voice, facial hair, general physical constitution) and estradiol is the main female sex hormone. It is responsible for development of secondary female characteristics and participates in the control of menstrual cycle. Progesterone is responsible for preparing the uterus for implantation of fertilised egg.
INTEXT QUESTION
Why cannot vitamin C be stored in our body?
What products would be formed when a nucleotide from DNA containing thymine is hydrolysed?
When RNA is hydrolysed, there is no relationship among the quantities of different bases obtained. What does this fact suggest about the structure of RNA?
-
Polymers.15
Do you think that daily life would have been easier and colourful without the discovery and varied applications of polymers? The use of polymers in the manufacture of plastic buckets, cups and saucers, children’s toys, packaging bags, synthetic clothing materials, automobile tyres, gears and seals, electrical insulating materials and machine parts has completely revolutionised the daily life as well as the industrial scenario. Indeed, the polymers are the backbone of four major industries viz. plastics, elastomers, fibres and paints and varnishes.
The word ‘polymer’ is coined from two Greek words: poly means many and mer means unit or part. The term polymer is defined as very large molecules having high molecular mass (103-107u). These are also referred to as macromolecules, which are formed by joining of repeating structural units on a large scale. The repeating structural units are derived from some simple and reactive molecules known as monomers and are linked to each other by covalent bonds. The process of formation of polymers from respective monomers is called polymerisation.
Classification of Polymers
There are several ways of classification of polymers based on some special considerations. One of the common classifications of polymers is based on source from which polymer is derived.
Under this type of classification, there are three sub categories.
1. Natural polymers
These polymers are found in plants and animals. Examples are proteins, cellulose, starch, some resins and rubber.
2. Semi-synthetic polymers
Cellulose derivatives as cellulose acetate (rayon) and cellulose nitrate, etc. are the usual examples of this sub category.
3. Synthetic polymers
A variety of synthetic polymers as plastic (polythene), synthetic fibres (nylon 6,6) and synthetic rubbers (Buna - S) are examples of man-made polymers extensively used in daily life as well as in industry.
Polymers can also be classified on the basis of their structure, molecular forces or modes of polymerisation.
Chain terminating step
For termination of the long chain, these free radicals can combine in different ways to form polythene. One mode of termination of chain is shown as under:
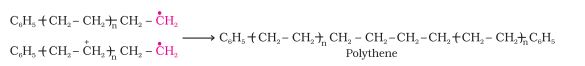
The addition polymers formed by the polymerisation of a single monomeric species are known as homopolymers, for example polythene discussed above is a homopolymer.
The polymers made by addition polymerisation from two different monomers are termed as copolymers. Buna-S, which is formed by polymerisation of buta–1, 3–diene and styrene is an example of copolymer formed by addition polymerisation.
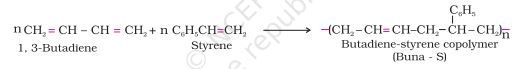
Polythene
Some Important Addition Polymers
Polythenes are linear or slightly branched long chain molecules. These are capable of repeatedly softening on heating and hardening on cooling and are thus thermoplastic polymers.
There are two types of polythene as given below:
1. Low density polythene: It is obtained by the polymerisation of ethene under high pressure of 1000 to 2000 atmospheres at a temperature of 350 K to 570 K in the presence of traces of dioxygen or a peroxide initiator (catalyst). The low density polythene (LDP) is obtained through the free radical addition and H-atom abstraction. It has highly branched structure. These polymers have straight chain structure with some branches as shown below.
Low density polythene is chemically inert and tough but flexible and a poor conductor of electricity. Hence, it is used in the insulation of electricity carrying wires and manufacture of squeeze bottles, toys and flexible pipes.
2.High density polythene: It is formed when addition polymerisation of ethene takes place in a hydrocarbon solvent in the presence of a catalyst such as triethylaluminium and titanium tetrachloride (Ziegler-Natta catalyst) at a temperature of 333 K to 343 K and under a pressure of 6-7 atmospheres. High density polythene (HDP) thus produced, consists of linear molecules as shown below and has a high density due to close packing. Such polymers are also called linear polymers. High density polymers are also chemically inert and more tough and hard. It is used for manufacturing buckets, dustbins, bottles, pipes, etc.
b. Polytetrafluoroethene (Teflon)
Teflon is manufactured by heating tetrafluoroethene with a free radical or persulphate catalyst at high pressures. It is chemically inert and resistant to attack by corrosive reagents. It is used in making oil seals and gaskets and also used for non – stick surface coated utensils.

c. Polyacrylonitrile
The addition polymerisation of acrylonitrile in presence of a peroxide catalyst leads to the formation of polyacrylonitrile/.

Polyacrylonitrile is used as a substitute for wool in making commercial fibres as orlon or acrilan.
EXAMPLE 1
Is
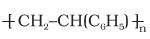 a homopolymer or a copolymer?
a homopolymer or a copolymer?SOLUTION
It is a homopolymer and the monomer from which it is obtained is styrene C6H5CH = CH2.
Condensation Polymerisation or Step Growth Polymerisation
This type of polymerisation generally involves a repetitive condensation reaction between two bi-functional or trifunctional mono-meric units. These polycondensation reactions may result in the loss of some simple molecules as water, alcohol, hydrogen chloride, etc., and lead to the formation of high molecular mass condensation polymers.In these reactions, the product of each step is again a bi-functional species and the sequence of condensation goes on. Since, each step produces a distinct functionalised species and is independent of each other, this process is also called as step growth polymerisation.
The formation of terylene or dacron by the interaction of ethylene glycol and terephthalic acid is an example of this type of polymerisation.
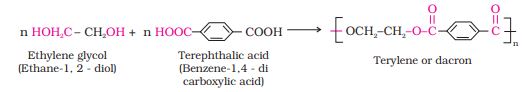
Some Important Condensation Polymers
a. Polyamides
These polymers possessing amide linkages are important examples of synthetic fibres and are termed as nylons. The general method of preparation consists of the condensation polymerisation of diamines with dicarboxylic acids or condensation of amino acids or their lactams.
Nylons
(1) Nylon 6,6: It is prepared by the condensation polymerisation of hexamethylenediamine with adipic acid under high pressure and at high temperature.
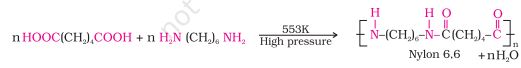
Nylon 6, 6 is fibre forming solid. It possess high tensile strength. This characteristic can be attributed to the strong intermolecular forces like hydrogen bonding. These strong forces also lead to close packing of chains and thus impart crystalline nature.
Nylon 6, 6 is used in making sheets, bristles for brushes and in textile industry.
(2) Nylon 6: It is obtained by heating caprolactum with water at a high temperature.
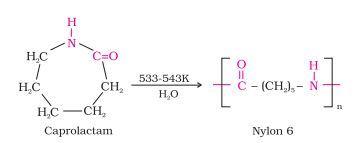
(b) Polyesters
These are the polycondensation products of dicarboxylic acids and diols. Dacron or terylene is the best known example of polyesters. It is manufactured by heating a mixture of ethylene glycol and terephthalic acid at 420 to 460 K in the presence of zinc acetate-antimony trioxide catalyst as per the reaction given earlier. Dacron fibre (terylene) is crease resistant and is used in blending with cotton and wool fibres and also as glass reinforcing materials in safety helmets, etc.(c) Phenol – formaldehyde polymer (Bakelite and related polymers)
Phenol – formaldehyde polymers are the oldest synthetic polymers. These are obtained by the condensation reaction of phenol with formaldehyde in the presence of either an acid or a base catalyst. The reaction starts with the initial formation of o-and/or p-hydroxymethylphenol derivatives, which further react with phenol to form compounds having rings joined to each other through–CH2 groups. The initial product could be a linear product – Novolac used in paints.
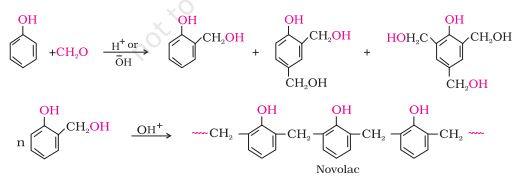
Novolac on heating with formaldehyde undergoes cross linking to form an infusible solid mass called bakelite. It is thermosetting polymer which cannot be reused or remoulded. Thus, bakelite is formed by cross linking of linear chains of the polymer novolac. Bakelite is used for making combs, phonograph records, electrical switches and handles of various utensils.
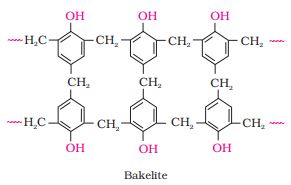
(d) Melamine — formaldehyde polymer
Melamine formaldehyde polymer is formed by the condensation polymerisation of melamine and formaldehyde.
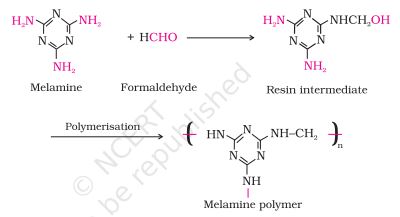
It is used in the manufacture of unbreakable crockery.
INTEXT QUESTION
Write the names of monomers of the following polymers:
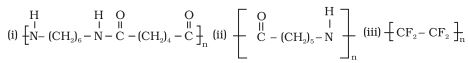
Classify the following as addition and condensation polymers: Terylene, Bakelite, Polythene, Teflon.
Copolymerisation
Copolymerisation is a polymerisation reaction in which a mixture of more than one monomeric species is allowed to polymerise and form a copolymer. The copolymer can be made not only by chain growth polymerisation but by step growth polymerisation also. It contains multiple units of each monomer used in the same polymeric chain.
For example, a mixture of buta–1, 3–diene and styrene can form a copolymer.
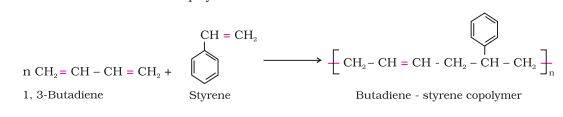
Copolymers have properties quite different from homopolymers. For example, butadiene - styrene copolymer is quite tough and is a good substitute for natural rubber. It is used for the manufacture of autotyres, floortiles, footwear components, cable insulation, etc.
Rubber
1. Natural rubber
Rubber is a natural polymer and possesses elastic properties. It is also termed as elastomeric polymer. In elastomeric polymers, the polymer chains are held together by the weak intermolecular forces. These weak binding forces permit the polymer to be stretched. A few ‘crosslinks’ are introduced in between the chains, which help the polymer to retract to its original position after the force is released.
Rubber has a variety of uses. It is manufactured from rubber latex which is a colloidal dispersion of rubber in water. This latex is obtained from the rubber tree which is found in India, Srilanka, Indonesia, Malaysia and South America.
Natural rubber may be considered as a linear polymer of isoprene (2-methyl-1, 3-butadiene) and is also called as cis - 1, 4 - polyisoprene.
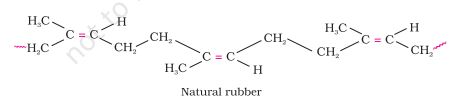
The cis-polyisoprene molecule consists of various chains held together by weak van der Waals interactions and has a coiled structure. Thus, it can be stretched like a spring and exhibits elastic properties.
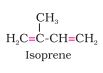
Vulcanisation of rubber: Natural rubber becomes soft at high temperature (>335 K) and brittle at low temperatures (<283 K) and shows high water absorption capacity. It is soluble in non- polar solvents and is non-resistant to attack by oxidising agents. To improve upon these physical properties, a process of vulcanisation is carried out. This process consists of heating a mixture of raw rubber with sulphur and an appropriate additive at a temperature range between 373 K to 415 K. On vulcanisation, sulphur forms cross links at the reactive sites of double bonds and thus the rubber gets stiffened.
In the manufacture of tyre rubber, 5% of sulphur is used as a crosslinking agent. The probable structures of vulcanised rubber molecules are depicted below:
2. Synthetic rubbers
Synthetic rubber is any vulcanisable rubber like polymer, which is capable of getting stretched to twice its length. However, it returns to its original shape and size as soon as the external stretching force is released. Thus, synthetic rubbers are either homopolymers of 1, 3 - butadiene derivatives or copolymers of 1, 3 - butadiene or its derivatives with another unsaturated monomer.
Preparation of Synthetic Rubbers
1.Neoprene
Neoprene or polychloroprene is formed by the free radical polymerisation of chloroprene.
It has superior resistance to vegetable and mineral oils. It is used for manufacturing conveyor belts, gaskets and hoses.
2. Buna – N
You have already studied about Buna-S, in Section 15.1.3. Buna
–N is obtained by the copolymerisation of 1, 3 – buta–1, 3–diene and acrylonitrile in the presence of a peroxide catalyst.

It is resistant to the action of petrol, lubricating oil and organic solvents. It is used in making oil seals, tank lining, etc.
INTEXT QUESTION
Explain the difference between Buna-N and Buna-S.
the following polymers in increasing order of their intermolecular forces. Nylon 6,6, Buna-S, Polythene.
Molecular Mass of Polymers
Polymer properties are closely related to their molecular mass, size and structure. The growth of the polymer chain during their synthesis is dependent upon the availability of the monomers in the reaction mixture. Thus, the polymer sample contains chains of varying lengths and hence its molecular mass is always expressed as an average. The molecular mass of polymers can be determined by chemical and physical methods.
Biodegradable Polymers
A large number of polymers are quite resistant to the environmental degradation processes and are thus responsible for the accumulation of polymeric solid waste materials. These solid wastes cause acute environmental problems and remain undegraded for quite a long time. In view of the general awareness and concern for the problems created by the polymeric solid wastes, certain new biodegradable synthetic polymers have been designed and developed. These polymers contain functional groups similar to the functional groups present in biopolymers.
Aliphatic polyesters are one of the important classes of biodegradable polymers. Some important examples are given below:
1. Poly \(\beta\)-hydroxybutyrate – co-\(\beta\)-hydroxy valerate (PHBV) It is obtained by the copolymerisation of 3-hydroxybutanoic acid and 3 - hydroxypentanoic acid. PHBV is used in speciality packaging, orthopaedic devices and in controlled release
of drugs. PHBV undergoes bacterial degradation in the environment.
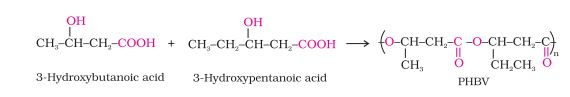
2. Nylon 2–nylon 6
It is an alternating polyamide copolymer of glycine (H2N–CH2– COOH) and amino caproic acid [H2N (CH2)5 COOH] and is biodegradable. Can you write the structure of this copolymer?
Polymers of Commercial Importance
Besides, the polymers already discussed, some other commercially important polymers along with their structures and uses are given below in Table 15.1.
Table 15.1: Some Other Commercially Important Polymers
-
Chemistry in Everyday life.16
By now, you have learnt the basic principles of chemistry and also realised that it influences every sphere of human life. The principles of chemistry have been used for the benefit of mankind. Think of cleanliness — the materials like soaps, detergents, household bleaches, tooth pastes, etc. will come to your mind. Look towards the beautiful clothes — immediately chemicals of the synthetic fibres used for making clothes and chemicals giving colours to them will come to your mind. Food materials — again a number of chemicals about which you have learnt in the previous Unit will appear in your mind. Of course, sickness and diseases remind us of medicines — again chemicals. Explosives, fuels, rocket propellents, building and electronic materials, etc., are all chemicals. Chemistry has influenced our life so much that we do not even realise that we come across chemicals at every moment; that we ourselves are beautiful chemical creations and all our activities are controlled by chemicals. In this Unit, we shall learn the application of Chemistry in three important and interesting areas, namely – medicines, food materials and cleansing agents.
Drugs and Their Classification
Drugs are chemicals of low molecular masses (~100 – 500u). These interact with macromolecular targets and produce a biological response. When the biological response is therapeutic and useful, these chemicals are called medicines and are used in diagnosis, prevention and treatment of diseases. Most of the drugs used as medicines are potential poisons, if taken in doses higher than those recommended. Use of chemicals for therapeutic effect is called chemotherapy.
Classification of Drugs
Drugs can be classified mainly on criteria outlined as follows:
a. On the basis of pharmacological effect
This classification is based on pharmacological effect of the drugs. It is useful for doctors because it provides them the whole range of drugs available for the treatment of a particular type of problem. For example, analgesics have pain killing effect, antiseptics kill or arrest the growth of microorganisms.
b. On the basis of drug action
It is based on the action of a drug on a particular biochemical process. For example, all antihistamines inhibit the action of the compound, histamine which causes inflammation in the body. There are various ways in which action of histamines can be blocked. You will learn about this in Section 16.3.2.
c. On the basis of chemical structure
It is based on the chemical structure of the drug. Drugs classified in this way share common structural features and often have similar pharmacological activity. For example, sulphonamides have common structural feature, given below.
Structural features of sulphonamides
d. On the basis of molecular targets
Drugs usually interact with biomolecules such as carbohydrates, lipids, proteins and nucleic acids. These are called target molecules or drug targets. Drugs possessing some common structural features may have the same mechanism of action on targets. The classification based on molecular targets is the most useful classification for medicinal chemists.
Drug-Target Interaction
Macromolecules of biological origin perform various functions in the body. For example, proteins which perform the role of biological catalysts in the body are called enzymes, those which are crucial to communication system in the body are called receptors. Carrier proteins carry polar molecules across the cell membrane. Nucleic acids have coded genetic information for the cell. Lipids and carbohydrates are structural parts of the cell membrane. We shall explain the drug-target interaction with the examples of enzymes and receptors.
Enzymes as Drug Targets
(a) Catalytic action of enzymes
For understanding the interaction between a drug and an enzyme, it is important to know how do enzymes catalyse the reaction (Section 5.2.4). In their catalytic activity, enzymes perform two major functions:
(1) The first function of an enzyme is to hold the substrate for a chemical reaction. Active sites of enzymes hold the substrate molecule in a suitable position, so that it can be attacked by the reagent effectively.
Substrates bind to the active site of the enzyme through a variety of interactions such as ionic bonding, hydrogen bonding, van der Waals interaction or dipole-dipole interaction (Fig. 16.1).
Fig. 16.1.
(a) Active site of an enzyme (b) Substrate (c) Substrate held in active site of the enzyme
(2) The second function of an enzyme is to provide functional groups that will attack the substrate and carry out chemical reaction.
b. Drug-enzyme interaction
Drugs inhibit any of the above mentioned activities of enzymes. These can block the binding site of the enzyme and prevent the binding of substrate, or can inhibit the catalytic activity of the enzyme. Such drugs are called enzyme inhibitors.
Drugs inhibit the attachment of substrate on active site of enzymes in two different ways;
(1) Drugs compete with the natural substrate for their attachment on the active sites of enzymes. Such drugs are called competitive inhibitors (Fig. 16.2).
Fig. 16.2. Drug and substrate competing for active site
(2) Some drugs do not bind to the enzyme’s active site. These bind to a different site of enzyme which is called allosteric site. This binding of inhibitor at allosteric site (Fig.16.3) changes the shape of the active site in such a way that substrate can- not recognise it.
Fig. 16.3: Non-competitive inhibitor changes the active site of enzyme after binding at allosteric site.
If the bond formed between an enzyme and an inhibitor is a strong covalent bond and cannot be broken easily, then the enzyme is blocked permanently. The body then degrades the enzyme-inhibitor complex and synthesises the new enzyme.
Receptors as Drug Targets
Receptors are proteins that are crucial to body’s communication process. Majority of these are embedded in cell membranes (Fig. 16.4). Receptor proteins are embedded in the cell membrane in such a way that their small part possessing active site projects out of the surface of the membrane and opens on the outside region of the cell membrane (Fig. 16.4).
Fig. 16.4 Receptor protein embedded in the cell membrane, the active site of the receptor opens on the outside region of the cell.
In the body, message between two neurons and that between neurons to muscles is communicated through certain chemicals. These chemicals, known as chemical messengers are received at the binding sites of receptor proteins. To accommodate a messenger, shape of the receptor site changes. This brings about the transfer of message into the cell. Thus, chemical messenger gives message to the cell without entering the cell (Fig. 16.5).
Fig. 16.5: (a) Receptor receiving chemical messenger
(b) Shape of the receptor changed after attachment of messenger
(c) Receptor regains structure after removal of chemical messenger.
There are a large number of different receptors in the body that interact with different chemical messengers. These receptors show selectivity for one chemical messenger over the other because their binding sites have different shape, structure and amino acid composition.
Drugs that bind to the receptor site and inhibit its natural function are called antagonists. These are useful when blocking of message is required. There are other types of drugs that mimic the natural messenger by switching on the receptor, these are called agonists. These are useful when there is lack of natural chemical messenger.Therapeutic Action of Different Classes of Drugs
In this Section, we shall discuss the therapeutic action of a few important classes of drugs.
Antacids
Over production of acid in the stomach causes irritation and pain. In severe cases, ulcers are developed in the stomach. Until 1970, only treatment for acidity was administration of antacids, such as sodium hydrogencarbonate or a mixture of aluminium and magnesium hydroxide. However, excessive hydrogencarbonate can make the stomach alkaline and trigger the production of even more acid. Metal hydroxides are better alternatives because of being insoluble, these do not increase the pH above neutrality. These treatments control only symptoms, and not the cause. Therefore, with these metal salts, the patients cannot be treated easily. In advanced stages, ulcers become life threatening and its only treatment is removal of the affected part of the stomach.
A major breakthrough in the treatment of hyperacidity came through the discovery according to which a chemical, histamine, stimulates the secretion of pepsin and hydrochloric acid in the stomach. The drug cimetidine (Tegamet), was designed to prevent the interaction of histamine with the receptors present in the stomach wall. This resulted in release of lesser amount of acid. The importance of the drug was so much that it remained the largest selling drug in the world until another drug, ranitidine (Zantac), was discovered.
Antihistamines
Histamine is a potent vasodilator. It has various functions. It
contracts the smooth muscles in the bronchi and gut and relaxes other muscles, such as those in the walls of fine blood vessels. Histamine is also responsible for the nasal congestion associated with common cold and allergic response to pollen.
Synthetic drugs, brompheniramine (Dimetapp) and terfenadine (Seldane), act as antihistamines. They interfere with the natural action of histamine by competing with histamine for binding sites of receptor where histamine exerts its effect.
Now the question that arises is, “Why do above mentioned antihistamines not affect the secretion of acid in stomach?” The reason is that antiallergic and antacid drugs work on different receptors.
Neurologically Active Drugs
(a) Tranquilizers
Tranquilizers and analgesics are neurologically active drugs. These affect the message transfer mechanism from nerve to receptor.
Tranquilizers are a class of chemical compounds used for the treatment of stress, and mild or even severe mental diseases. These relieve anxiety, stress, irritability or excitement by inducing a sense of well-being. They form an essential component of sleeping pills. There are various types of tranquilizers. They function by different mechanisms. For example, noradrenaline is one of the neurotransmitters that plays a role in mood changes. If the level of noradrenaline is low for some reason, then the signal-sending activity becomes low, and the person suffers from depression. In such situations, antidepressant drugs are required. These drugs inhibit the enzymes which catalyse the degradation of noradrenaline. If the enzyme is inhibited, this important neurotransmitter is slowly metabolised and can activate its receptor for longer periods of time, thus counteracting the effect of depression. Iproniazid and phenelzine are two such drugs.
Some tranquilizers namely, chlordiazepoxide and meprobamate, are relatively mild tranquilizers suitable for relieving tension. Equanil is used in controlling depression and hypertension.
Derivatives of barbituric acid viz., veronal, amytal, nembutal, luminal and seconal constitute an important class of tranquilizers. These derivatives are called barbiturates. Barbiturates are hypnotic, i.e., sleep producing agents. Some other substances used as tranquilizers are valium and serotonin.
b. Analgesics
Analgesics reduce or abolish pain without causing impairment of consciousness, mental confusion, incoordination or paralysis or some other disturbances of nervous system. These are classified as follows:
1. Non-narcotic (non-addictive) analgesics
2. Narcotic drugs
1. Non-narcotic (non-addictive) analgesics: Aspirin and paracetamol belong to the class of non-narcotic analgesics. Aspirin is the most familiar example. Aspirin inhibits the synthesis of chemicals known as prostaglandins which stimulate inflammation in the tissue and cause pain. These drugs are effective in relieving skeletal pain such as that due to arthritis. These drugs have many other effects such as reducing fever (antipyretic) and preventing platelet coagulation. Because of its anti blood clotting action, aspirin finds use in prevention of heart attacks.
2. Narcotic analgesics: Morphine and many of its homologues, when administered in medicinal doses, relieve pain and produce sleep. In poisonous doses, these produce stupor, coma, convulsions and ultimately death. Morphine narcotics are sometimes referred to as opiates, since they are obtained from the opium poppy.
These analgesics are chiefly used for the relief of postoperative pain, cardiac pain and pains of terminal cancer, and in child birth.

Antimicrobials
Diseases in human beings and animals may be caused by a variety of microorganisms such as bacteria, virus, fungi and other pathogens. An antimicrobial tends to destroy/prevent development or inhibit the pathogenic action of microbes such as bacteria (antibacterial drugs), fungi (antifungal agents), virus (antiviral agents), or other parasites (antiparasitic drugs) selectively. Antibiotics, antiseptics and disinfectants are antimicrobial drugs.
(a) Antibiotics
The search for chemicals that would adversely affect invading bacteria but not the host began in the nineteenth century. Paul Ehrlich, a German bacteriologist, conceived this idea. He investigated arsenic based structures in order to produce less toxic substances for the treatment of syphilis. He developed the medicine, arsphenamine, known as salvarsan. Paul Ehrlich got Nobel prize for Medicine in 1908 for this discovery. It was the first effective treatment discovered for syphilis. Although salvarsan is toxic to human beings, its effect on the bacteria, spirochete, which causes syphilis is much greater than on human beings. At the same time, Ehrlich was working on azodyes also. He noted that there is similarity in structures of salvarsan and

The structures of salvarsan, prontosil azodye and sulphapyridine showing structural similarity.
azodyes. The –As = As– linkage present in arsphenamine resembles the –N = N – linkage present in azodyes in the sense that arsenic atom is present in place of nitrogen. He also noted tissues getting coloured by dyes selectively. Therefore, Ehrlich began to search for the compounds which resemble in structure to azodyes and selectively bind to bacteria. In 1932, he succeeded in preparing the first effective antibacterial agent, prontosil, which resembles in structure to the compound, salvarsan. Soon it was discovered that in the body prontosil is converted to a compound called sulphanilamide, which is the real active compound. Thus the sulpha drugs were discovered. A large range of sulphonamide analogues was synthesised. One of the most effective is sulphapyridine.H.W. Florey and Alexander Fleming shared the Nobel prize for Medicine in 1945 for their independent contributions to the development of penicillin.
Despite the success of sulfonamides, the real revolution in antibacterial therapy began with the discovery of Alexander Fleming in 1929, of the antibacterial properties of a Penicillium fungus. Isolation and purification of active compound to accumulate sufficient material for clinical trials took thirteen years.
Antibiotics have either cidal (killing) effect or a static (inhibitory) effect on microbes. A few examples of the two types of antibiotics are as follows:
Bactericidal Bacteriostatic
Penicillin Erythromycin
Aminoglycosides Tetracycline
Ofloxacin Chloramphenicol
The range of bacteria or other microorganisms that are affected by a certain antibiotic is expressed as its spectrum of action. Antibiotics which kill or inhibit a wide range of Gram-positive and Gram-negative bacteria are said to be broad spectrum antibiotics. Those effective mainly against Gram-positive or Gram-negative bacteria are narrow spectrum antibiotics. If effective against a single organism or disease, they are referred to as limited spectrum antibiotics. Penicillin G has a narrow spectrum. Ampicillin and Amoxycillin are synthetic modifications of penicillins. These have broad spectrum. It is absolutely essential to test the patients for sensitivity (allergy) to penicillin before it is administered. In India, penicillin is manufactured at the Hindustan Antibiotics in Pimpri and in private sector industry.
Chloramphenicol, isolated in 1947, is a broad spectrum antibiotic. It is rapidly absorbed from the gastrointestinal tract and hence can be given orally in case of typhoid, dysentery, acute fever, certain form of urinary infections, meningitis and pneumonia. Vancomycin and ofloxacin are the other important broad spectrum antibiotics. The antibiotic dysidazirine is supposed to be toxic towards certain strains of cancer cells.
(b) Antiseptics and disinfectants
Antiseptics and disinfectants are also the chemicals which either kill or prevent the growth of microorganisms.
Antiseptics are applied to the living tissues such as wounds, cuts, ulcers and diseased skin surfaces. Examples are furacine, soframicine, etc. These are not ingested like antibiotics. Commonly used antiseptic, dettol is a mixture of chloroxylenol and terpineol. Bithionol (the compound is also called bithional) is added to soaps to impart antiseptic properties. Iodine is a powerful antiseptic. Its 2-3 per cent solution in
alcohol-water mixture is known as tincture of iodine. It is applied on wounds. Iodoform is also used as an antiseptic for wounds. Boric acid in dilute aqueous solution is weak antiseptic for eyes.
Disinfectants are applied to inanimate objects such as floors, drainage system, instruments, etc. Same substances can act as an antiseptic as well as disinfectant by varying the concentration. For example, 0.2 per cent solution of phenol is an antiseptic while its one percent solution is disinfectant.
Chlorine in the concentration of 0.2 to 0.4 ppm in aqueous solution and sulphur dioxide in very low concentrations, are disinfectants.
Antifertility Drugs
Antibiotic revolution has provided long and healthy life to people. The life expectancy has almost doubled. The increased population has caused many social problems in terms of food resources, environmental issues, employment, etc. To control these problems, population is required to be controlled. This has lead to the concept of family planning. Antifertility drugs are of use in this direction. Birth control pills essentially contain a mixture of synthetic estrogen and progesterone derivatives. Both of these compounds are hormones. It is known that progesterone suppresses ovulation. Synthetic progesterone derivatives are more potent than progesterone. Norethindrone is an example of synthetic progesterone derivative most widely used as antifertility drug. The estrogen derivative which is used in combination with progesterone derivative is ethynylestradiol (novestrol).

INTEXT QUESTION
Sleeping pills are recommended by doctors to the patients suffering from sleeplessness but it is not advisable to take its doses without consultation with the doctor. Why ?
With reference to which classification has the statement, “ranitidine is an antacid” been given?
Chemicals in Food
Chemicals are added to food for (i) their preservation, (ii) enhancing their appeal, and (iii) adding nutritive value in them. Main categories of food additives are as follows:
1. Food colours
2. Flavours and sweeteners
3. Fat emulsifiers and stabilising agents
4. Flour improvers - antistaling agents and bleaches
5. Antioxidants
6. Preservatives
7. Nutritional supplements such as minerals, vitamins and amino acids.
Except for chemicals of category (vii), none of the above additives have nutritive value. These are added either to increase the shelf life of stored food or for cosmetic purposes. In this Section we will discuss only sweeteners and food preservatives.
Artificial Sweetening Agents
Natural sweeteners, e.g., sucrose add to calorie intake and therefore many people prefer to use artificial sweeteners. Ortho-sulphobenzimide, also called saccharin, is the first popular artificial sweetening agent. It has been used as a sweetening agent ever since it was discovered in 1879. It is about 550 times as sweet as cane sugar. It is excreted from the body in urine unchanged. It appears to be entirely inert and harmless when taken. Its use is of great value to diabetic persons and people who need to control intake of calories. Some other commonly marketed artificial sweeteners are given in Table 16.1.
Table 16.1: Artificial Sweeteners
Aspartame is the most successful and widely used artificial sweetener. It is roughly 100 times as sweet as cane sugar. It is methyl ester of dipeptide formed from aspartic acid and phenylalanine. Use of aspartame is limited to cold foods and soft drinks because it is unstable at cooking temperature.
Alitame is high potency sweetener, although it is more stable than aspartame, the control of sweetness of food is difficult while using it. Sucralose is trichloro derivative of sucrose. Its appearance and taste are like sugar. It is stable at cooking temperature. It does not provide calories.
Food Preservatives
Food preservatives prevent spoilage of food due to microbial growth. The most commonly used preservatives include table salt, sugar, vegetable oils and sodium benzoate, C6H5COONa. Sodium benzoate is used in limited quantities and is metabolised in the body. Salts of sorbic acid and propanoic acid are also used as preservatives.
INTEXT QUESTION
Why do we require artificial sweetening agents ?
Antioxidants in Food
These are important and necessary food additives. These help in food preservation by retarding the action of oxygen on food. These are more reactive towards oxygen than the food material which they are protecting. The two most familiar antioxidants are butylated hydroxy toluene (BHT) and butylated hydroxy anisole (BHA). The addition of BHA to butter increases its shelf life from months to years.
Sometimes BHT and BHA along with citric acid are added to produce more effect. Sulphur dioxide and sulphite are useful antioxidants for wine and beer, sugar syrups and cut, peeled or dried fruits and vegetables.
Cleansing Agents
In this Section, we will learn about detergents. Two types of detergents are used as cleansing agents. These are soaps and synthetic detergents. These improve cleansing properties of water. These help in removal of fats which bind other materials to the fabric or skin.
Soaps
Soaps are the detergents used since long. Soaps used for cleaning purpose are sodium or potassium salts of long chain fatty acids, e.g., stearic, oleic and palmitic acids. Soaps containing sodium salts are formed by heating fat (i.e., glyceryl ester of fatty acid) with aqueous sodium hydroxide solution. This reaction is known as saponification.In this reaction, esters of fatty acids are hydrolysed and the soap obtained remains in colloidal form. It is precipitated from the solution by adding sodium chloride. The solution left after removing the soap contains glycerol, which can be recovered by fractional distillation. Only sodium and potassium soaps are soluble in water and are used for cleaning purposes. Generally potassium soaps are soft to the skin than sodium soaps. These can be prepared by using potassium hydroxide solution in place of sodium hydroxide.
Types of soaps
Basically all soaps are made by boiling fats or oils with suitable soluble hydroxide. Variations are made by using different raw materials.
Toilet soaps are prepared by using better grades of fats and oils and care is taken to remove excess alkali. Colour and perfumes are added to make these more attractive.
Soaps that float in water are made by beating tiny air bubbles before their hardening. Transparent soaps are made by dissolving the soap in ethanol and then evaporating the excess solvent.
In medicated soaps, substances of medicinal value are added. In some soaps, deodorants are added. Shaving soaps contain glycerol to prevent rapid drying. A gum called, rosin is added while making them. It forms sodium rosinate which lathers well. Laundry soaps contain fillers like sodium rosinate, sodium silicate, borax and sodium carbonate.
Soap chips are made by running a thin sheet of melted soap onto a cool cylinder and scraping off the soaps in small broken pieces. Soap granules are dried miniature soap bubbles. Soap powders and scouring soaps contain some soap, a scouring agent (abrasive) such as powdered pumice or finely divided sand, and builders like sodium carbonate and trisodium phosphate. Builders make the soaps act more rapidly. The cleansing action of soap has been discussed in Unit 5.
Why do soaps not work in hard water?
Hard water contains calcium and magnesium ions. These ions form insoluble calcium and magnesium soaps respectively when sodium or potassium soaps are dissolved in hard water.
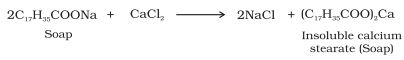
These insoluble soaps separate as scum in water and are useless as cleansing agent. In fact these are hinderance to good washing, because the precipitate adheres onto the fibre of the cloth as gummy mass. Hair washed with hard water looks dull because of this sticky precipitate. Dye does not absorb evenly on cloth washed with soap using hard water, because of this gummy mass.
Synthetic Detergents
Synthetic detergents are cleansing agents which have all the properties of soaps, but which actually do not contain any soap. These can be used both in soft and hard water as they give foam even in hard water. Some of the detergents give foam even in ice cold water.
Synthetic detergents are mainly classified into three categories:
1. Anionic detergents (ii) Cationic detergents and (iii) Non-ionic detergents
(1) Anionic Detergents: Anionic detergents are sodium salts of sulphonated long chain alcohols or hydrocarbons. Alkyl hydrogensulphates formed by treating long chain alcohols with concentrated sulphuric acid are neutralised with alkali to form anionic detergents. Similarly alkyl benzene sulphonates are obtained by neutralising alkyl benzene sulphonic acids with alkali.
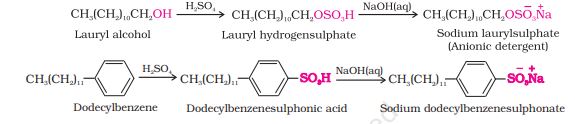
In anionic detergents, the anionic part of the molecule is involved in the cleansing action. Sodium salts of alkylbenzenesulphonates are an important class of anionic detergents.
They are mostly used for household work. Anionic detergents are also used in toothpastes.
(2) Cationic Detergents: Cationic detergents are quarternary ammonium salts of amines with acetates, chlorides or bromides as anions. Cationic part possess a long hydrocarbon chain and a positive charge on nitrogen atom. Hence, these are called cationic detergents. Cetyltrimethylammonium bromide is a popular cationic detergent and is used in hair conditioners.
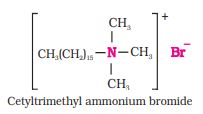
Cationic detergents have germicidal properties and are expensive, therefore, these are of limited use.
(3) Non-ionic Detergents: Non-ionic detergents do not contain any ion in their constitution. One such detergent is formed when stearic acid reacts with polyethyleneglycol.
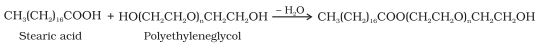
Liquid dishwashing detergents are non-ionic type. Mechanism of cleansing action of this type of detergents is the same as that of soaps. These also remove grease and oil by micelle formation.
Main problem that appears in the use of detergents is that if their hydrocarbon chain is highly branched, then bacteria cannot degrade this easily. Slow degradation of detergents leads to their accumulation. Effluents containing such detergents reach the rivers, ponds, etc. These persist in water even after sewage treatment and cause foaming in rivers, ponds and streams and their water gets polluted.
These days the branching of the hydrocarbon chain is controlled and kept to the minimum. Unbranched chains can be biodegraded more easily and hence pollution is prevented.
INTEXT QUESTION
Write the chemical equation for preparing sodium soap from glyceryl oleate and glyceryl palmitate. Structural formulae of these compounds are given below.
1. (C15H31COO)3C3H5 – Glyceryl palmitate
2. (C17H32COO)3C3H5 – Glyceryl oleate
Following type of non-ionic detergents are present in liquid detergents, emulsifying agents and wetting agents. Label the hydrophilic and hydrophobic parts in the molecule. Identify the functional group(s) present in the molecule.